AnaK
-
Постов
30 -
Зарегистрирован
-
Посещение
Достижения AnaK
")





-
Спасибо за вашу помощь и комментарии Ну не осадок, а наночастицы сформированные из коллоида...:D Я не знаю что это. Интересный момент: я определила концентрацию вещества (которую изначально кладу), свыше которой я начинаю наблюдать этот эффект повышения концентрации после фильтрации. Также, чем больше начальная концентрация тем больше я получаю повышение в концентрации после фильтрации. Такое возможно если растворитель каким то образом не проходит через фильтр (а растворитель вполне нормальная не вязкая жидкость, например диэтилсульфоксид), а вещество проходит (в форме мелких наночастиц - из литературы они могут формироваться, размер от 100 до 800 микрон?). Фильтр 0.22 микрона PTFE.... Вы думаете что эффект не полного пика в ГХ может наблюдаться только в случае когда они вместе с растворенным веществом ? Потому что в контроле без вещества количество полученное из хроматограммы хорошо бьется с начальным. Максимальная ошибка 5%. Кроме того, я попробовала второй метод и он дал то же самое повышение концентрации: навеска вещества растворена в растворителе, фильтрация, фильтрат взвешивается и помещается в вакуумную печь (45С, 40мбар) - через сутки растворитель испарился, осталось только вещество. Его вес меряю, считаю - опять растворимость больше максимально возможной изначально. Т.е. на мой взгляд проблема не в стадии ГХ, а в стадии фильтрации, но не понятно в чем именно.
-
Может ли быть такое, что формируются наночастицы вещества, которые идут через фильтр (0.22 мкм) и в то же время не являются растворенными. Параллельно с этим, растворитель как-то застревает на фильтре (потому что изначальный раствор супер концентрирован, выглядит как гель)?
-
Да, вы все верно поняли. Вещество В нелетучее и неопределяемое на ГХ. Оно скорее всего остается в септуме при инжектировании или размазывается в начале колонки...ее кончик (точнее начало) потом можно отрезать 😄 Причины разбавления фильтрата с помощью ДМСО: 1. малое количество фильтрата (0.05г недостаточное количество для образца, надо в чем то растворить, а больше сделать не получается - уж больно ценное вещество В) 2. очень большая вязкость фильтрата, выглядит практически как гель! Такую ерунду вкалывать тоже не особо здорово (вся игла уделается). Проблема еще и в том, что растворитель кипит свыше 200С и так просто его не удалить. Конечно можно было бы подключить вакуум и температуру, но это много мороки для 0.05г и таких образцов еще около 20... Поэтому придумала такой метод, а теперь пытаюсь понять адекватен ли он. Это да....да вот только у меня растворители нетривиальные совсем, некоторые твердые при комнатной температуре, данным по ним нет совершенно, кое-как в принципе удалось найти эти химикаты. Более того вещество В очень крупное, скорее как небольшой органический полимер из 20 звеньев, нежели соль.
-
Я использовала ГХ-ПИД и делала калибровку по растворителю (в ДМСО), в 5 концентрациях и двух независимых повторах. Пробовала делать контроль без вещества: только растворитель пропускаю через фильтр, разбавляю, меряю - показывает количество растворителя с ошибкой 5%. Другой контроль: без фильтрования вещества (и изначально точно отвешенными массами и в расчете на то что абсолютно все растворилось): метод дает массу растворителя с ошибкой 4%, количество растворенного вещества с ошибкой 9%, в конце концов ошибка значения растворимости около 12%. Но эти 12% все же не объясняют результаты.
-
да, вы правы что изначальная концентрация 0.25 г/г. Стоит заметить, что понятие "растворимость" немного отличается: это масса растворенного вещества деленная на массу растворителя (не раствора). Так возможно ли такое, что после фильтрации концентрация увеличивается? По идее это физически невозможно? Почему то я получаю значение растворимости выше, чем изначальная концентрация. Конкретные данные: Изначально кладется 0.1 г вещества и 0.3г растворителя. Фильтрат (0.22 мкм фильтр): 0.15г. ДМСО: 1.07г. Масса растворителя в фильтрате (из хроматограммы): 0.091г. Итого, масса растворенного вещества в фильтрате должна быть: 0.15-0.091=0.059. Таким образом, растворимость вещества равна 0.059/0.091=0.648 г/г. Как такое возможно если изначально максимальная концентрация 0.25 г/г? Где может быть проблема?
-
Добрый день! Подскажите пожалуйста, как объяснить странные результаты определения растворимости (валидны ли они с физико-химической точки зрения)? Мне нужно померить растворимость вещества (В) в данном растворителе (Р). План: приготовить перенасыщенный раствор, отфильтровать и померить количество в фильтрате. Я кладу в емкость 1г (В) и 3г (Р). Максимально возможная концентрация вещества (если все растворилось) 1/3=0.333 г/г. Все перемешивается. Далее я фильтрую какое-то количество раствора, так что не растворенное вещество не может пройти. У меня получается фильтрат известной массы (например 2г), в который я добавляю ДМСО для разбавления (известное количество, например 5г), затем я в этом конечном растворе определяю количество растворителя (Р) (!не вещества!) с помощью ГХ. В итоге, чтобы узнать количество вещества в фильтрате я вычитаю из общей массы раствора (5+2=7г) количество ДМСО и количество растворителя (определенное ГХ, например 1г). Получается 7-5-1=1г вещества в фильтрате. Получаю растворимость вещества 1г (В) / 1г (Р) = 1 г/г. Может ли такое быть если изначально максимально возможная концентрация была 0.333г? То есть может ли получаться растворимость вещества по этой методике больше чем изначальная концентрация?
-
Нам интересно получить рибозу из ксилозы, т.к. мы изначально используем лигноцеллюлозное сырье, где почти нет рибозы, а вот ксилозы очень много.
-
Спасибо за информацию, звучит действительно очень толково! Я думаю, что как-то возможно сделать так, чтобы защита была только справа и тогда у нас один гидроксил первичный, а другой вторичный и нужно селективно протозилировать по вторичному гидроксилу, верно? А может быть можно депротектировать этот сахар частично (только левую часть)? Или, например, применить какие-то другие защитные группы, которые снимаются по-разному (разными реагентами). Может быть использовать два разных типа и затем отсеить молекулы у которых одна сторона защищена одним типом, а другая другим? Хотя это звучит сложно, наверняка выход будет совсем небольшой, если и получится...
-
Изменение конформации связи в сахарах синтетическим путем
AnaK опубликовал тема в Органическая химия
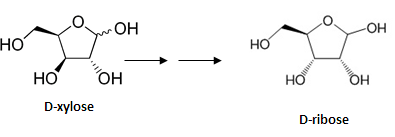
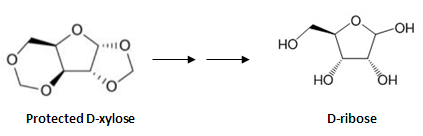
Добрый день, форумчане! Возник вопрос, касающийся сахаров: возможно ли каким-либо образом превратить Д-ксилозу в Д-рибозу (желательно синтетическим методом)? Т.е. по сути нужно изменить конформацию одной связи. Подскажите, пожалуйста, возможно ли это? И также, возможно ли это сделать из защищенной ксилозы (2 картинка ниже), которая имеется у нас в большом количестве (защитная группа может быть модифицирована)? Я не спец в органической химии, ни разу не видела подобных примеров в учебниках.
-
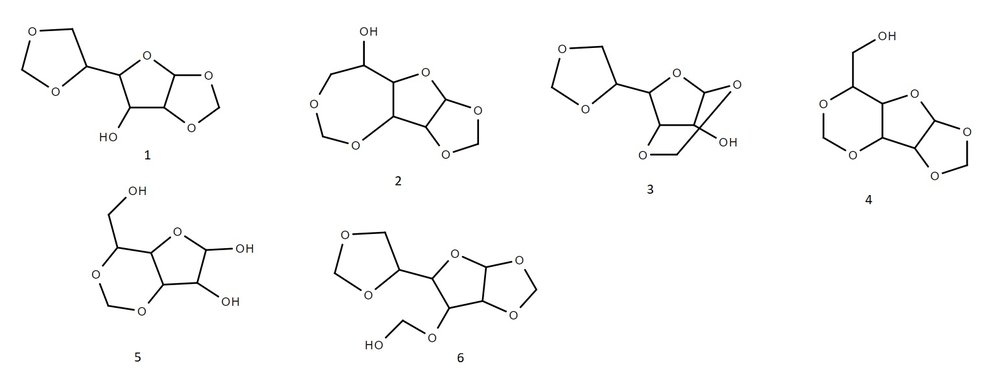
Есть смесь различных изомеров (см. картинку), при этом вещества 1 и 4 значительно преобладают в количестве. Не подскажете ли методы как можно селективно провести превращение OH группы ПЕРВИЧНЫХ спиртов в H (чтобы в итоге там осталась торчать только метил группа)? Возможно ли сначала провести какое-нибудь замещение OH только в первичных спиртах на более хорошую уходящую группу, а потом восстановить? Как потом их отделить от непрореагировавших вторичных соединений? Условия также должны быть достаточно мягкими, чтобы сохранить ацетали (т.е. без воды и без кислоты).
-
Еще такая особенность, эти защищенные глюкозы плохо растворяются в органике (из можно более менее сносно растворить в смеси метанола и этилацетата) и все равно забирают воду из атмосферы, и непонятно как исключить попадание воды в реакционную смесь.
-
Нашла несколько статей, где с ним проделывали дегидратацию спиртов, не разрушая ацетали. Но оказалось, что этот реагент достаточно токсичен и его использование сопряжено с рядом трудностей, вроде хранения в инертной атмосфере, а также то что он очень влагочувствителен, дорогой и не может быть получен из возобновляемых источников. Нет ли более безопасного пути, например использования какого-нибудь катализатора? Вы что-нибудь об этом может быть знаете?
-
Не особо. Вообще эти вещества при гидролизе с кислотой Бронстеда идут обратно в сахара, а потом уже в фурфурол и т.д. Просто возникла мысль, может быть из них что-то сделать. Так называемый upgrading
-
Эти защищенные сахара были получены из биомассы изначально. Если обрабатывать биомассу формальдегидом, то получается в 3-7 раза увеличить выходы сахаров (cellulose and hemicellulose) и лигнина по сравнению с процессами, не включающими обработку формальдегидом. Это связано с тем, что защитные ацетальные группы препятствуют образованию C-C связей в ходе обработки и экстракции. Т.е. по сути, мы по умолчанию получаем вот такие вот молекулы из глюкозы, когда разделяем фракции изначальной биомассы. Дальше вопрос: а как их использовать. Данный процесс обработки формальдегидом был описан 2-3 года назад в журнале Science и на данный момент это один из лучших способов обработки биомассы.
-
Спасибо Вам большое!!! Я подробно изучу это и, возможно, в скором времени даже попробую его использовать.