

lexa_xto_802
-
Постов
101 -
Зарегистрирован
-
Посещение
Тип контента
Профили
Форумы
События
Сообщения, опубликованные lexa_xto_802
-
-
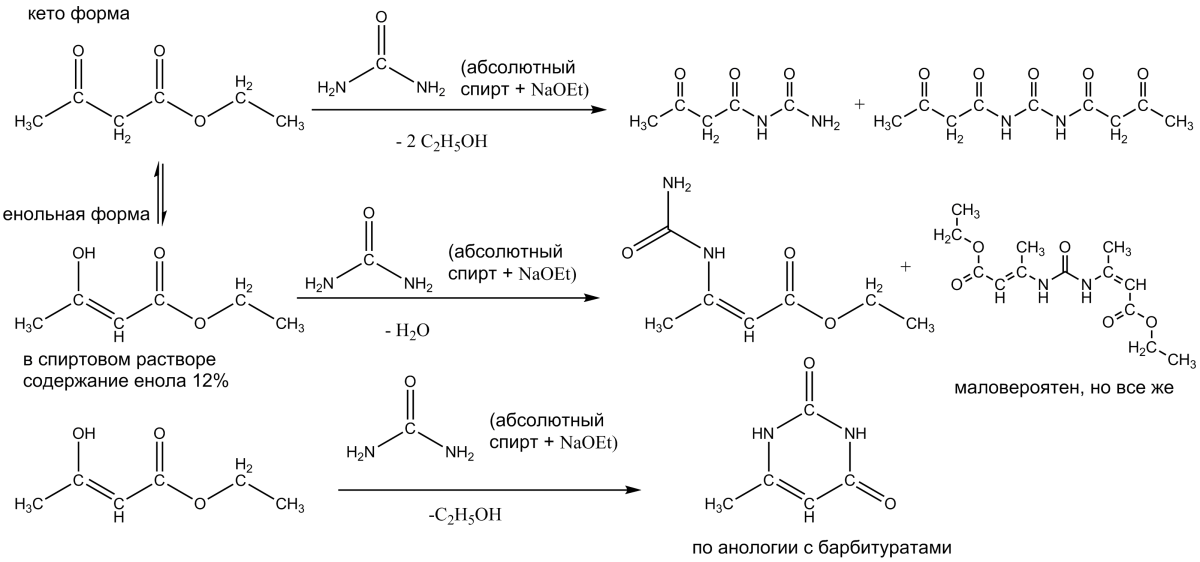
Всех приветствую, особенно экспертов органического синтеза!
Вопрос следющий, какой механизм наиболее вероятен?

Конечно есть еще несколько вариантов гипотетических (н-р амидный эфир енольной формы и т.д.) но изображать не стал..
-
Вы что-нибудь про "суперселективные фазы" слышали?
Да слышал и видел как работают, НО увы не имею в наличии...
Прилагаю методику которую нашел на просторах интернета, обратите внимание на пики 19, 20
Нужно проверить, рабочая или нет в моих условиях...
-
А с чего вы решили, что у вас они не поделились?
Потому что делал последовательно анализ, сначала бензол вкалывал снимал хроматограмму, потом в туда же добавлял циклогексан и обратно снимал хроматограмму (хроматограмм с собой нет поэтому не могу привести пример)
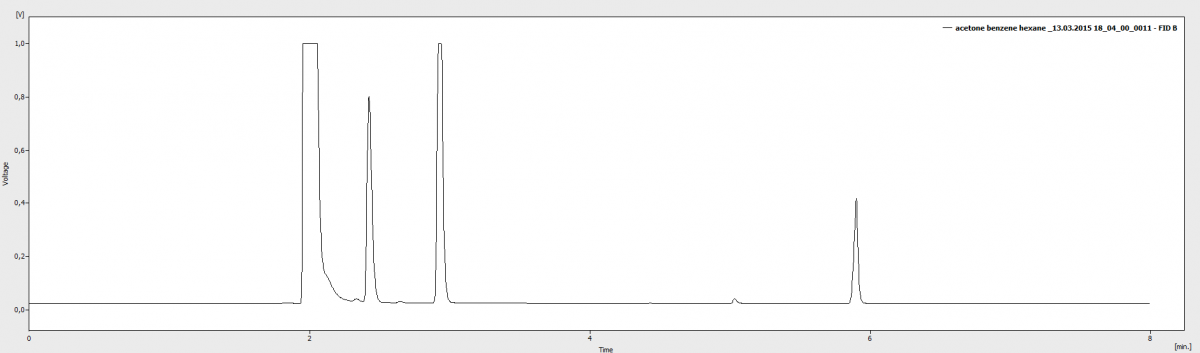
На второй картинке смесь состоит из ацетона (1 пик) гексан (2 пик) бензол+циклогексан (3 пик) циклогексанон (4 пик), веществ 5 а пиков 4
Вещества добавлялись последовательно...
думаю из-за того что бензол кипит при 80,1 С а циклогексан при 80,74 С (а в большинстве случаев но не во всех, на этой колонке выходят вещества примерно по температурам кипения) их сложновато разделить
-
Сквалан - это алкан С30Н62, по строению тритерпен.
Извиняюсь это моя ошибка, сквалан это НННЕЕ 100% метилсиликон как писал ранее... посмотрел в справочнике.
А выходят они чётко в один пик или с плечиками?
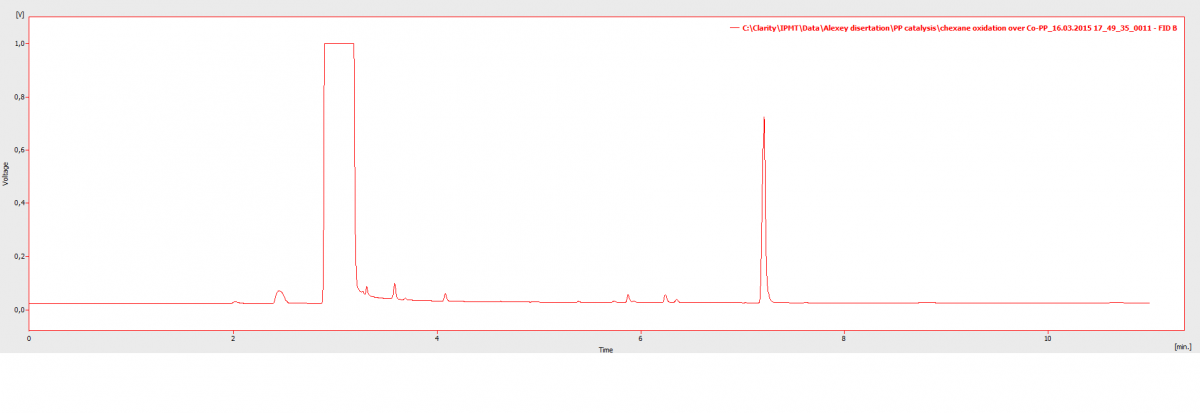
Бензол то у меня растворитель, само собой он в перегрузе (рис. 1)
Делал смесь бензола и циклогексана в другом растворителе, всеравно выходят одним пиком (рис. 2) 3 пик (немного в перегрузе но это так сказать тестовый закол)
Хроматограммы получены с помощью разных методик, т.е. не идентичные условия
-
А есть колонка со скваланом?
К великому сожалению не имеется(((
По сути сквалан это 100% % метилсиликон (каучук), у меня же в наличии почти тоже самое только 5% финила и 95% метилсиликон (каучук) т.е. чуть более полярная... по идее разница не слишком большая должна быть, но на практике ничего не выходит.
А какой длины, диаметр, толщина фазы Вы порекомендуете колонку со скваланом ???
-
Всем доброго времени суток!
Сразу к делу...
Есть газовый хроматограф Dani Master GC (Италия),
Колонка DN-5 ((5% Phenyl) - 95% methylpolysiloxane), уже 27,8 м (было когда то 30 м
), диаметр 0,25 мм, толщина фазы 0,45 мкм.Анаголичные колонки с той же фазой для сравнения это
007-2, CP-Sil 8CB, DB TM -5, DB TM -5.625, HP TM -5, SAC-5, OV TM -5, PTE-5, PTE-5QTM, PAS-5, RSL-200, Rtx TM -5, SE-54, SPB-5, ULTRA-2, XTI-5, SE-52, BP-5, PE-2, ZB-5, AT TM -5, EC TM -5
Инжектор split/splitless (Т 200 С)Детектор FID (ПИД, пламеноионизационный) (Т 300 С)Шприц Хэмильтон 10 мкл, автосэмплер, ввод образца от 0,5 до 1 мклСобственно суть проблемы... не могу разделить бензол и циклогексан выходят одним пиком.Помогите создать/подобрать методику...P.S. в литературе конечно есть данные как разделить эту смесь на данной колонке, НО там длинна 60 м, диаметр 0,32 мм, фаза 1 мкм... пробовал по этой методике у меня не разделились -
Меня грызут сомнения вообще в целесообразности/эффективности метода синтеза эфиров ацетоуксусной кислоты методом переэтерификации
Просто есть ацетоуксусный эфир, ацетоуксусной кислоты нет (синтезировать на тот момент не хотелось). Нужно повесть подлиннее гидрофобный хвост для определенных целей.
СН кислотность группы СН2 не так сильно выражена, но конечно у альфа углеродного атома карбонильной группы есть заместитель СООС2Н5, что в свою очередь увеличивает СН-кислотность этой самой СН2 группы, но все же соглашусь с товарищем yatcheh "далеко не достаточна для того, шобы напрочь загасить алкоголят".
Так что будем дальше проводить переэтерификацию... только теперь думаю конц. серной или акоголятом... ???
До этого проводил подобную реакцию с этими же веществами тогда шибко не запаривался, на выход не смотрел... после реакции загасил все водой (нахрен сделал? сам не помню), избавился от кислоты (а тогда я аж 1 мл конц налил вместо в данном случае 0,2, да и первый опыт с малым количеством делал). В итоге при комнатной температуре этот сложный эфир "замерз" хлопьями, проэкстрагировал бензолом, отогнал бензол... получил что надо и сделал с ним свое коварное дело, дальше пустил в синтез и там все сработало... Теперь нужно побольше количество "наделать" так как не все исследования успел сделать... Вот и думаю как сделать 100% по спирту ...
Может быть еще долить АУЭ добавить заново конц серной и погреть... или всетаки алкоголят добавить?
-
Попробуйте катионит КУ-2 (правда, в нём воды 15%). Его и извлекать нетрудно - фильтрованием
Для переэтерификации этилового эфира АУК метанолом я брал натрий (предварительно растворял в метаноле). Где-то полпроцента (насколько помню).
Алкоголяты на переэтерификацию спиртами работают НАМНОГО лучше, чем кислоты, не раз в этом убеждался.
Катионит свою воду наверное никому не отдаст... нашел в продаже КУ-2-8, попробуем позже...
Алкоголят докозанола наверное проблематично получить, еще не пробовал, но думаю при сильном нагревании только возможно, это не метиловый спирт((( или может быть в растворителе попрет...
-
Реакционная масса не доварена! Посему я бы добавил хороший избыток АУЭ, катализатор и погрел бы до тех пор, пока не исчез спирт. После этого убираем катализатор и в вакууме отгоняем избыточный АУЭ (остатки можно удалить азеотропно с ксилолм, кажется). Полужидкий остаток - продукт-сырец, если повезёт, то, м.б., закристаллизуется "из себя" или из гексана.
Да хроматограф показывает что недоварена.
Делал так, при Т90С соотношение АУЭ с докозанолом 4/1 по молям, 0,2мл конц серной кислоты как катализатор. В ротационном испарителе 4-5 часов на скорости 100 об/мин с очень слабым вакуумом чтобы спирт этиловый моментально улетал. Но оказывается что недоваривается в таких условиях...
Какой катализатор Вы посоветуете и как его извлечь? и какой избыток АУЭ бы Вы посоветовали? или может быть поменять условия реакции...?
А что именно мешает - этилацетоацетат, или докозанол?
Можно было бы избытком этилацетоацетата связать весь докозанол (с отгонкой этанола), а потом на вакууме при хорошем нагревании отжать этиловый эфир. Весь он, конечно не отожмётся (для этого надо докозаноловый эфир довести до кипения), но до какого-то минимального количества его содержание можно довести.
Нужен чистый эфир докозонил ацетоацетат... Докозанол отогнать и убрать никак, так как он везде будет растворяться вместе и эфиром докозонилАУЭ, экстракция получается не вариант.
АУЭ (этилацетоацетат) еще можно в вакууме на ротационном отогнать, с ним я разберусь... а докозанол-1 вот мешает сильно...
-
Всем доброго времени суток! Нужен совет...
Есть смесь, состоящая из: этиловый эфир ацетоуксусной кислоты, докозанол-1 (С22Н45ОН), и продукт их совместной переэтерификации докозонил ацетоацетат... Как и чем их можно разделить. (перегонка не идет даже под вакуумом т.к. докозанол-1 кипит 180С при 0,22 мм рт.с., а такой вакуум у меня нет возможности создать)
Нужна очень срочно помощь!
-
просмотрите Органикум, по моему Т2.
Спасибо большое! Отличная память у Вас! на странице 90 нашел что искал!
Всем удачи!
-
Всем доброго времени суток!
Подскажите пожалуйста, есть кто-нибудь кто проводил реакции ацилирования спиртов хлорангидридами карбоновых кислот? Где можно почерпнуть прописи? С какими трудностями вы сталкивались (кроме образующегося HCl). Проводили ли вы реакцию с добавлением пиридина, и при каких условиях...
Необходимо решить подобную практическую задачу...
Всем удачи!
-
Всем здравствуйте!
Просьба о помощььи полимерщиков-синтетиков. Помогите (пнуть в нужном направлении, подсказать литературу, помочь методичкой) решить задачу по синтезу поли-а-олефинов. Желательно помочь методикой (прописью).
Из реактивов есть: а-олефины, AlCl3, FeCl3, TiCl4 (старенький).
Пробовал по методике (прикрепленный файл) не вышло, может быть кто знаком с методикой синтеза полимеров катионной полимеризацией?
-
Так тебе что нужно? "упрочнить" (сделать крепче) так сказать или просто высушить я так и не понял, в чем задача?
Если просто хочешь чтобы не трескалось попробуй сушить в вакууме (если сильный вакуум можешь даже почти без нагрева 30-35С достаточно), должно помочь! Проблем с испарением воды не будет так как это более менее деликатный вариант сушки (лиофилизация)...
А если вообще хочешь чтобы ничего не изменилось в структуре, нужно заморозить мокрый материал и тоже в вакууме сушить, только потребуется мощный вакуумный насос с большим объемом перекачки и без нагревания...
Вот такое у меня соображение.
-
Рабек Я. Экспериментальные методы в химии полимеров, часть 1, 2 будут очень полезны!!!! в этих книгах не помню в какой части есть описание этого метода.
Суть заключается в использовании растворителя в котором не растворяется полимер но растворяются мономеры и прочее... Так сказать "хороший" и "плохой" растворитель. Я эту методику называю "хороший-плохой полицейский")))). Добавляешь осторожно, по каплям, к раствору растворитель в котором не растворяется полимер и видишь как раствор начнет мутнеть (это выпадают крупинки полимера), потом отфильтровать, собрать и еще разок можно промыть. потом сушишь и несешь порошочек на анализ.
Есть на кафедре полимерщики? Они точно в курсе что нужно делать, обратись за советом...
Возьми 5-6 разных ТИПОВ!!! растворителей и проведи экспресс тест. колбочки по 1 мл растворителя и добавляй по 1 КАПЛЕ раствора полимера, посмотри где выпадет осадок (помутнеет колбочка). можешь поискать литературу где что из твоих мономеров растворяется. Вариантов масса...
Просто так для интереса можете не отвечать. Это простой "студенческий" опыт? или работа (дипломная/кандидатская и т.д.)?
-
Нужно попробовать ароматику (бензол или толуол) и полярные растворители, так сказать провести экспресс тест на растворимость в колбочках например 5-6 проб думаю достаточно чтобы охватить весь спектр наиболее часто встречающихся растворителей.
-
 2
2
-
-
Lexa_xto, спасибо Вам огромное за рекомендации!
я в надежде, что реакция пройдет и без активации, смешала полимеры с водой и продержала их при 100 градусах в течении 1,5 часов. раствор получился чистый, прозрачный, невязкий.
теперь вот понесу на ИК.
только вот не знаю, какую концентрацию им приготовить...
Попробуйте переосадить полученный полимер, многократно промыть от непрореагировавшего jeffamine. Не вижу смысла нести смесь так как она есть, разделить. Потом порошок запрессовать с бромидом калия (я пользуюсь соотношением 150 мг KBr и 5 мг полимера). Водные растворы на ИК никто не даст анализировать
-
если бы перед Вами стояла такая задача?
Скорее всего конденсация пойдет как с простыми первичными аминами, но с небольшим выходом... из-за размеров молекул... Похожую реакцию встречал в статье, там тоже малеиновый ангидрид (уже в полимерной форме) конденсировали с первичными аминами если память не изменяет... посмотри может быть в ссылках есть что-то полезное...
В списках лит-ры должно быть что-то похожее. Еще активно использую сайт orgsyn.org там вообще все синтезы есть и все методики рабочие проверенные...
Если представить ваш jeffamine как R-NH2 то вот например можно посмотреть на это http://orgsyn.org/demo.aspx?prep=CV5P0944 и не смотрите что статейка 1961 года она 100% должна быть рабочая, в любом случае пробуйте.
Удачи!
Synthesis of polymeric pour point depressants for Nada crude.pdf
-
1
-
-
Да с посудой у меня все норм! Я ведь пиротехник)! Как же я сразу не додумался про измерение плотности!!)))))))))) вот я ид..т!!!!!!!!!!!!!!! Спс)!!
Ну ну пиротехник... Хоть как спорьте без нормальной посуды и четкого соблюдения методики, без каких либо "ай да и так сойдет", ничего нормального не получится. Температура очень важна... Чистый все равно не получишь, получишь фракцию от 36 до 40 кипения...
Что там речь шла про топливо, а не проще купить нитрометан для моделей или на крайняк изооктан (растворитель) (ОЧ=100)
http://rcshop.kz/product/1135/ (цена на 5 делить)
-
Здравствуйте Всем!
К делу...
Необходимо получить СН2=СН-С(О)ОR R=C22H45. Помогите поиском методики есть вот такое http://orgsyn.org/demo.aspx?prep=CV3P0146 . В принципе метода устраивает, но хотелось бы почитать что-нибудь еще (язык рус англ не важен).
У кого какие идеи.
-
Всем все сразу станет понятно как и что
http://vk.com/himik_psihopat?z=video211076308_168182889%2F534d594ef223fc4547
-
Уважаемые форумчане помогите найти книгу Вигдергауз М.С. Расчеты в газовой хроматографии.
-
Уважаемые форумчане помогите найти книгу Вигдергауз М.С. Расчеты в газовой хроматографии.
-
Уважаемые форумчане помогите найти книгу Вигдергауз М.С. Расчеты в газовой хроматографии.
 ), диаметр 0,25 мм, толщина фазы 0,45 мкм.
), диаметр 0,25 мм, толщина фазы 0,45 мкм.
Ацетоуксусный эфир и Мочевина
в Органическая химия
Опубликовано
А кето форма амиды необразует? всетаки кетоформа занимает 88%. Вообще думается мне что будет образовываться смесь веществ по первому и последнему вариантам