
fastique
-
Постов
65 -
Зарегистрирован
Тип контента
Профили
Форумы
События
Сообщения, опубликованные fastique
-
-
што
Ты скитлс рассасываешь, наверно? Ты в основном просто механически слизистую повреждаешь конфетами, имхо
-
Такой вопрос, может ли салициловая кислота образовывать соответствующий фенолят при реакции с избытком водного раствора NaOH?
Всегда казалось, что реакция NaOH с салициловой к-той заканчивается на образовании соли (по карбоксильной группе), но не знаю почему, вот сейчас призадумался.
-
21 минуту назад, yatcheh сказал:
Для эффективного - не хватит.
Дефлегматор - фигня. "Эффективно" подразумевает насадочную колонку и головку полной конденсации. А с вашим дефлегматором, если у вас хватит терпения отбирать по капле в минуту - ну, тогда какое-то разделения можно добиться.
Хочется хотя бы минимум 96% чистоты толуольной фракции получить. Ничего кроме дефлегматора для разделения нет
 Есть еще один на 20 см, можно их вместе будет поставить
Есть еще один на 20 см, можно их вместе будет поставить -
-
А можно бром посушить P2O5? И с него же и отогнать?
-
А в петролейном эфире 70-100°С гексан есть? Всё-таки он (гексан) при 68°С кипит.
-
А сколько примерно времени занимает перегонка литра жидкости (к примеру, этилацетата)? А то я сегодня часа 2.5-3 потратил где-то, чувствую, что-то не правильно делал.
-
Подскажите, вот йодистый метилен (CH2I2) при хранении разлагается на какие продукты? Понятно, что на свободный иод, а ещё на что?
-
Чем дегазацию/нейтрализацию бромпропана можно сделать по-быстрому? Аммиак сойдет?
-
Можно ли использовать мол. сита 4А для сушки ДМСО? Если да, то увеличится ли время осушки по сравнению с ситами 3А?
-
А в каком виде обычно NaI продаётся? Дигидрат или б/в? На упаковке ничего не написано кроме "НАТРИЙ ИОДИСТЫЙ". Т.к. он в комках и мокрый, я предполагаю, что это NaI*2H2O.
И как его обезводить, если это дигидрат?
-
Вопрос остался актуальным...
-
При каких условиях проводить гидролиз сложного эфира? Конкретно - метилгептаноата (метилэнантата). Какой избыток щёлочи (KOH) брать, какой концентрации раствор щелочи делать, как долго кипятить и т.п.?
-
-
Написал в ЛС.
-
Сульфат железа 2 пролежал ~год на полке и стал полностью коричневого цвета. Видимо упаковка была не совсем герметичная. Вопрос: во что он превратился и как его обратно превратить в сульфат железа 2?
-
60% холодной отмывают. Бутанол кстати ведь в воде растворим заметно, высшие насколько-то дольше мыть придётся. А концентрированная серная сама сложный эфир переэтерифицирует.
Ну некоторые эфиры бутанола слабо, но растворимы в воде - большие потери эфира при хорошей промывке водой будут. А высшие спирты (октанол тот же) фиг отмоешь водой. Мыл октилпропионат примерно по литру воды раз 6, если не больше, задолбался знатно (самого эфира кстати около 10 мл) и всё равно запах октанола остался, и проба с дихроматом в 50% серной (после перегонки эфира с паром и еще одной промывки водой) даёт довольно сильную черную окраску.
-
Они с ней реагируют, образуя моноэфиры серной в равновесии с исходными в-вами.
Ну а реагируют смешиваясь же?
Мне это нужно только чтобы отмывать сложный эфир от спирта.Сейчас нашёл в "Vogel. Practical Organic Chemistry" кое-что. Там бутилбутират 7 раз по 5мл промывается 60% серной кислотой для удаления бутанола...
-
-
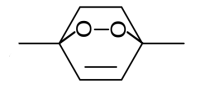
Думается, что нереально именно в таком виде. Чтобы Виттиг прошел, нужны мощные основания, которые сдепротонируют спирт, а там уже побочные реакции пойдут. Если только через защиту спирта. Кроме того, такой продукт (он кстати неверно нарисован), стоит 100% дешевле, чем исходный 4-бромбутанол.
Ну картинку с вики брал. Я протупил немного, мне нужен 3-бромпропанол для этой реакции же. Тут чисто спортивный интерес. Хочется синтезировать его самому
в небольших количествах... -
Добавить воды и отогнать с азеотропом. через дефлегматор, поддерживая минимальную температуру на головке, чтобы только слегка капало, одна капля в несколько секунд
Так азеотроп (октанол-вода) при 99.4 °С кипит. Там уже эфир с паром гнаться будет же?
А конкретно про идею с марганцовкой получится? Ну то есть сложный эфир не окислится марганцовкой?
-
Недавно открыл для себя эту реакцию и вот назрел вопрос.Реально ли синтезировать цис-3-гексенол по реакции Виттига? Вот как мне думается: 4-бромбутанол реагирует с трифенилфосфином, затем получившийся (4-гидрокси)трифенилфосфониум бромид реагирует с пропионовым альдегидом и получается сабж.
-
К примеру октилпропионат. Сделал его, используя большой избыток кислоты (2.5 эквивалента), т.к. Дина-Старка нет. Промыл пару раз содой, 5 раз тёплой водой (~300-350мл). Постороний запах октанола еще чувствуется. Вот как от него избавиться? Пришла идея окислить его марганцовкой в щелочном р-ре (Na2CO3) - получится? С эфиром ничего не случится?
-
Вот делаю я значит эфир какого-нибудь жирного спирта очень слабо растворимого в воде. Допустим октилформиат. Беру большой избыток муравьиной кислоты и долго нагреваю с н-октанолом в присутствии кат. количеств H2SO4. Потом промываю водой, р-р соды 3 раза, 1-2 раза горячей водой и перегоняю с паром. Как мне определить остался ли спирт (н-октанол в данном случае) в эфире в значительном количестве (больше 1%)? Дихромат калия в серной + пару капель исследуемого эфира поможет? Или недостаточно чувствительная реакция для таких количеств?
Конкретно про октилформиат я уже нашел решение проблемы: октанол жгучий на вкус, а октилформиат почти безвкусный.
Но вопрос остается актуальным.
 Есть еще один на 20 см, можно их вместе будет поставить
Есть еще один на 20 см, можно их вместе будет поставить 
 в небольших количествах...
в небольших количествах...
Синтез изопропилового спирта
в Органическая химия
Опубликовано
Восстановлением ацетона боргидридом :D