

Электрон
-
Постов
81 -
Зарегистрирован
-
Посещение
Тип контента
Профили
Форумы
События
Сообщения, опубликованные Электрон
-
-
-
Какая версия?
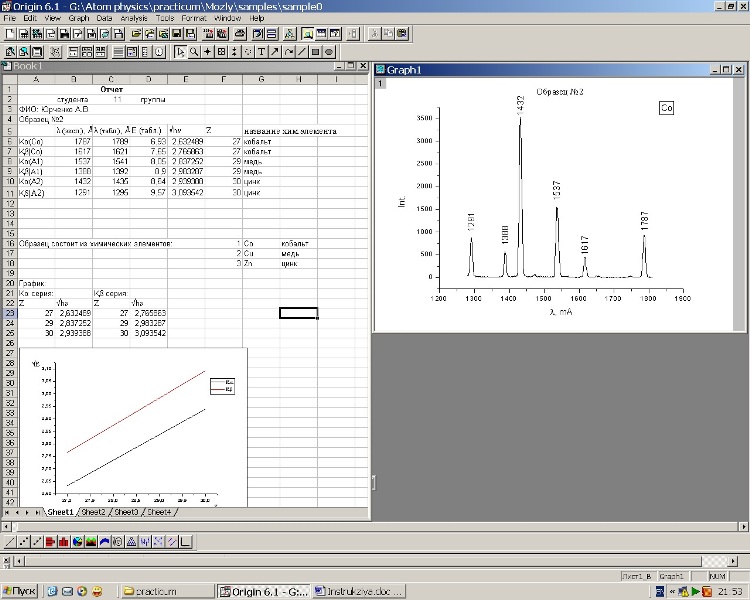
Слева на панели инструментов я красным обвёл инструмент "Text tool" - нажимаете на него, затем на область на графике, где нужна надпись, после чего выбираете пишете что Вам надо. Надпись можно редактировать, перемещать, менять шрифт. Если надо кириллица - выбираете Arial CUR
Версия программы 8.5.
С этой командой (Text tool), я разобрался, но что интересно не догадался применить её)). Действительно так можно подписать пики. Но скажите как такой текст перевернуть на 90 градусов?
Я нашел в интернете какой то пример работы в программе, и там приведен пример, который прикрепил ниже. Вот я так же хотел пики подписать.
Но это все работа в ручную. Я думаю что в программе должна быть команда, при помощи которой, я курсором навожу на точку графика и получаю данные в этой точки, которые сразу над ней или рядом появляются.

-
Здравствуйте.
Ребята, такая проблема. Нарисовал график рентгенограммы в программе Origin, и не могу проставить там подписи пиков. Хочу сделать так что бы было видно при каких углах пики. Не могу понять как подписать точку. Тыкал тыкал, получилось подписать шкалу Y, но это интенсивность, а мне нужны углы.
-
Добрый день. Во вкладке Phase option есть параметр Scale factor для каждой фазы. По умолчанию - 1. Меняя этот параметр для каждой фазы отдельно можно изменить соотношение фаз.
По второму вопросу - координат там нет. Потому да - искать в других источниках.
Все понял. Разобрался в программке, понял как делать, спасибо! По второму вопросу тоже ясно.
Проконсультируйте еще))
1. Вот пытаюсь я обнаружить рефлексы вещества в рентгенограмме. Сколько вообще минимум должно быть рефлексов, что бы можно было сказать что, да, это вещество есть в образце который исследовали?
2. В карточках, по крайне мере в тех что мне дали (одна из них прикреплена в сообщении выше), сразу указаны интенсивности и углы, причем некоторые строки (точнее в d. ang. ) выделены жирным, о чем это говорит? Что это самые главные рефлексы, и если они присутствуют то это точно это вещество.
-
... А экспериментальная дифрактограмма загружается из меню Diffraction - Load powder pattern.
З.Ы. У меня 2.3 (просто привычка). То что слетают настройки - ничего страшного, файл pwc.cfg неправильно прописывает при некоторых ошибках. Вариант - перераспаковать програму поверху или потереть этот файл - создаст новый. Диффрактограмма просто линией патамушта смотрите от какого до какого угла построена - там просто пиков нет.
Алексей здравствуйте.
Подскажите пожалуйста по программе PowderCell.
Пытаюсь расшифровать диффрактограммы, загружаю экспериментальную диффрактограмму и пару соединений которые там ищу. Загруженные соединения отображаются в процентном соотношении, если их два то по 50%, пишется, если три то по 33% и так далее. Скажите есть ли в программе возможность менять это процентное соотношение при загрузке веществ?
Еще проконсультируйте по карточкам РФА.
Мне дали карточки, но по некоторым есть вопрос. Прикрепил к сообщению карточку карбонат гидроксида лантана. В карточке указана пространственная группа, размеры ячейки, НО нету данных по расположению атомов в молекуле. Я не вижу эти данные или их тут и правда нет? Или тут как то по другому надо вносить данные в программу.
Если нет координат атомов то я ни чего ведь не построю? Тогда что делать? Искать другую карточку, в других местах.
-
ОСНОВНОЙ СПИСОК
(остатки на 22.06.2016)
Здравствуйте.
Хотим купить у Вас РЗЭ. Пару вопросов:
1. Вы можете выслать счет на покупку? Мы организация и работаем через счета. Или у вас только за наличный расчет?
2. Вы отправляете почтой России, проблем ни каких не возникало, все таки хим. вещества?
3. Для начала, нас интересуют следующие вещества:
Лютеция хлорид 6-водный - 20 грамм.
Гадолиния оксид ГдО-Л (99,9%) - 100 грамм.
Тербия сульфат 8-водный - 20 грамм.
В таком количестве есть вещества?
4. Ориентировочно сколько будет стоить доставка до Томска.
-
Вот тут проблема. Файл со структурой Вы загружаете правильно. А экспериментальная дифрактограмма загружается из меню Diffraction - Load powder pattern.
З.Ы. У меня 2.3 (просто привычка). То что слетают настройки - ничего страшного, файл pwc.cfg неправильно прописывает при некоторых ошибках. Вариант - перераспаковать програму поверху или потереть этот файл - создаст новый. Диффрактограмма просто линией патамушта смотрите от какого до какого угла построена - там просто пиков нет.
Ну теперь все получилось. Я не понял что надо было в другом месте делать загрузку. Теперь все грузится и строится.
Большое Вам спасибо!
И с диффрактограммой теперь понятно, все строится снова.
Еще раз, большое спасибо!
-
Очень странно, но у меня так ни чего и не получается.
Давайте сверимся версиями программ. У меня установлена версия 2.4 (8.03.2000).
При запуске сразу выскакивает сообщение о том, что надо использовать точку для разделения, а не запятую (Uses decimal points and not points commas...).
Далее я загружаю файл с построенным в программе гидроксидом лантана. А потом уже файл х_у. Оказывается что я не могу увидеть эти файлы! Это расширение не поддерживает программа.
Вчера я пытался загрузить файл с расширением txt. Я брал файл с расширением dat, чистил его как Вы сказали, удалял лишние единички (правда пробелы оставлял, об этой тонкости я не догадался
)) и пытался грузануть файл (поменяв его расширение в винраре). Сейчас до меня дошло что надо расширение х_у делать. (Прошлое Ваше сообщение не внимательно просмотрел). Захожу через архиватор, меняю расширение с txt, а программа не видит такой файл! Я пытаюсь открыть файл с расширением txt, файл подчистил, все лишнее удалил, но он не открывается, программа ругается. Сначала она пишет: Символ не найден. Используйте настройку 1. (Symbol not found. PowderCell use Set up 1.). Закрываю это окно, и выскакивает вторая ошибка, там вообще по немецкому написано: недопустимая операция с плавающей точкой. (Ungultige Gleitkommaoperation.)
Я подумал что у меня виндоус восьмерка, а программа явно на более ранние версии виндоус рассчитана, залез в свойства и поставил галочку совместимости. Проблеме это не помогло, но теперь у меня не строится график рентгенограммы! Загружаю гидроксид лантана, сохраненный, сама молекула строится, а в окне графика тупо прямая линия.
Капец, целая история с освоением этой программы)) я так понимаю где то я не то что то делаю.
-
Это координаты расположения атомов в элементарной ячейке кристаллической решетки... врядли стоит от гидроокиси лантана требовать молекулярной решетки.
Положите хденить файлы, я бы глянул... хотя если Эксель подтягивает, смените расширение с dat/raw на x_y... вопщем в файле дифрактограммы (подтягивается после построения теоретической, Load - Powder pattern) должны быть:
угол - интенсивность
угол - интенсивность
угол - интенсивность
угол - интенсивность
угол - интенсивность
угол - интенсивность
угол - интенсивность...
итд... и ничего больше.
З.Ы. Странно, что при удалении координат ничего не строит, должно бы строить, но с случайной интенсивностью пиков.
Про расположения атомов понятно, спасибо.
Загрузил присланные файлы на яндекс диск. Ссылка.
В архиве две папки.
Папка номер 1, это те файлы о которых ведется переписка в теме на форуме.
Папка номер 2, это анализ сделанный в другом месте, и там несколько другие присланные файлы.
Я пытался сначала переименовать расширение файлов в папке номер 1, как Вы написали, но ни чего у меня не вышло, файл не загружается, программа ругается и вообще глючит, и даже не грузит потом вообще ни чего пока её не перезапустишь.
Делал я все как Вы написали, сначала загрузил сохраненный файл с построенными пиками, а потом уже пытался грузить файл с "реальными" данными. И ни чего не грузится.
Потом начал рыться и вспомнил что в другое место отдавал образец на анализ, это папка номер два. Там кстати сравнение делали как раз в программе Powder Cell как я понял. И в этих присланных файлах уже имеются данные для построения, конкретно есть файл с расширением x_y. Но и его то же не удается загрузить. Программа ругается и все. Завтра еще попробую.
При удалении координат действительно строится рентгенограмма, я просто не обратили на это внимания, только на всплывшее сообщение об ошибке и все.
По идеи да, но РФА этого не показал. Видимо не много его там образовалось. Где то я слышал что РФА видеть начинает когда примеси более 1-2%, значит в пробе меньше))
-
1. Данные по структуре я взял в 12 посте. Если бы его не было - пришлось бы искать - литература, базы данных итд...
2. Рентген "видит" собственно не атомы, а электроны, т.е Лантан - сгусток из 57 электронов, кислород - из 8 с хвостиком... водород - "кучка" из 1 электрона, причем он смещен к кислороду и формирует этот самый "хвостик". Т.е. по честному, положение водорода по данным рентгеновской дифракции определить трудновато, да и на интенсивности пиков он повлияет не сильно.
З.Ы. Попробуйте вообще вытереть координаты атомов и посмотреть что будет (оно матюкнется типа "Нет координат", нажмите Ок).
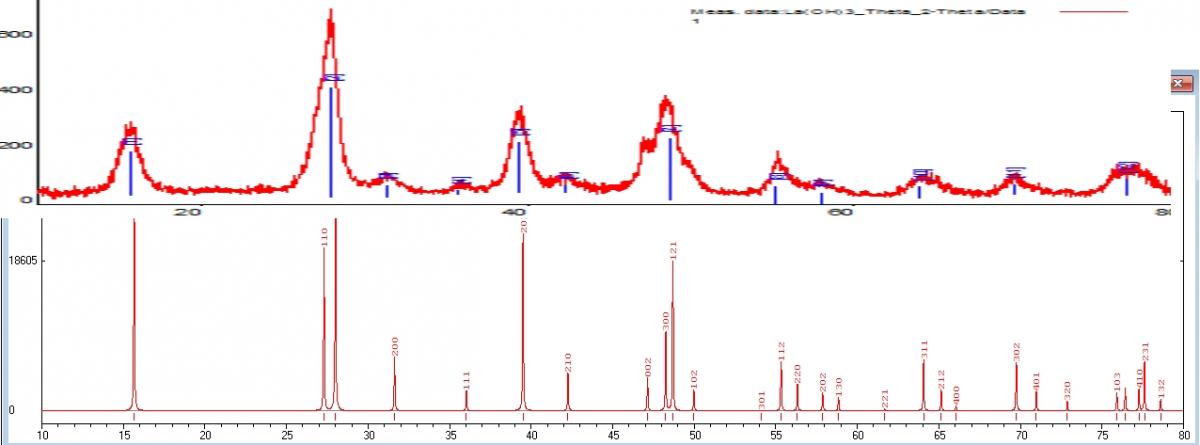
3. Вобщем, да, уширение пиков связано с напряжениями в кристаллах и с мелким размером кристаллов. Если отжечь, пики станут острее. Правда тут еще играют роль настройки дифрактометра и параметры съемки дифрактограммы (у Вас она т.н. "обычного качества" - т.е. для идентификации/фазового анализа хватит, для структурных исследований надо бы выдержку в точке побольше)
4. Да, так и есть - Одни теоретические (которые из справочника), а другие это экспериментальные и рассчитаны конкретно для Вашего образца.
З.Ы.2 Поищите, может у Вас есть собственно файл данных рентгенограммы - т.е. тот по которым можно экспериментальную рентгенограмму построить. Если есть, его можно подгрузить в Powder, он посчитать умеет (правда, не очень точно, зато в один клик).
1. Понятно, то есть в карточке для конкретного соединения должна быть указана информация по конкретному атому в молекуле. Кстати, я правильно понимаю что эти данные это координаты расположения атомов в молекуле?
2. Все понял, спасибо. Попробовал убрать координаты - программа ни чего не строит))
3. Все понятно. Я синтезировал гидроксид лантана из нитрата лантана с добавлением гидроксида натрия. Выпавший осадок простоял где то сутки, после чего я его фильтровал в центрифуге и отмывал от избытка гидроксида натрия. Кристаллы получились не важные и не совершенные. А вот если бы подержать подольше осадок, да еще подогреть его, то кристаллики получше бы были. Ну или подогреть уже высушенный гидроксид градусов до 150-200, то энергии уже будет побольше для кристалла и атомы перестроятся в более правильную позицию. С условиями съемки то же понятно.
По файлам для программы, я не смог ни чего загрузить. В файлах которые мне прислали расширения нужного нет.
Есть файлы с расширением RAS, dat и RAW.
Из файлов RAS и dat, научился с помощью блокнота вытаскивать координаты и строить в экселе график отдельный.
Может можно как то отдельно загружать координаты в программу Powder Cell, или нужен конкретный специальный файл для этого? Или просто в наглую расширение у dat файла поменять на txt и грузить в программу?
-
Еще раз очень всем благодарен за помощь!
Aversun спасибо за ссылку на программу, но я в Powder Cell пробую разобраться.
Маленко освоил Powder Cell, по крайне мери смог построить дифрактограмму и внести параметры для построения.
Прикрепляю изображения того что у меня получилось.
Проясните несколько вопросов:
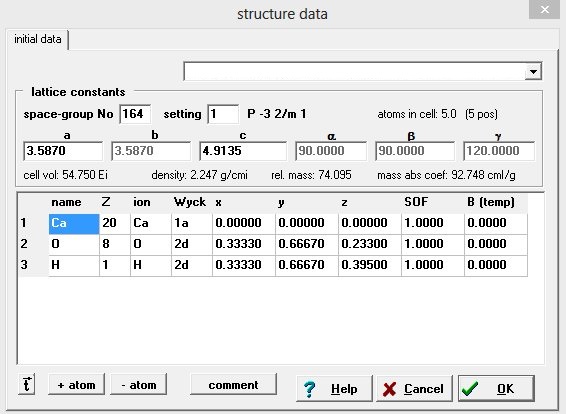
1. Леша гальваник, Вы объяснил что надо ввести только некоторые параметры и дальше все программа рассчитает. Действительно, ввел параметры и все рассчитано было самой программой. Нашел кнопочку в программе (HKL-List) где показаны углы и расстояния. Все так же как и у Вас в посте. Но я не понял где Вы взяли данные по самим атомам, обвел на рисунке (ниже прикреплен к письму) красным прямоугольником и цифрой один. Это какие то справочные данные и они стандартны?
2. То что построено в программе это и есть гидроксид лантана? В данных структуры, которые вносятся в ручную, указаны только лантан и кислород (цифра два на картинке) (я вводил по примеру Леши гальваника), но по идеи молекула записана как La(OH)3, это надо как то пояснять в программе, что там еще и водород, и их по три атома?
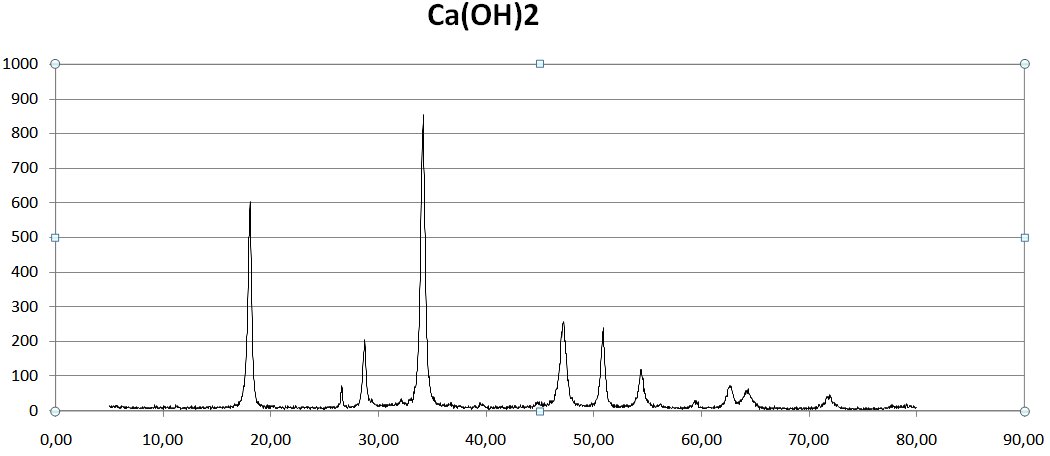
3. Программа построила дифрактограмму. На рисунке который прикрепил к сообщению сравнение между реальными данными и расчетными. Верхний график реальный, нижний расчетный. Специально растянул что бы было по шкалам совпадения. (рисунок корявый, сделал для вопроса). Как видно на реальном графике рефлексы не такие острые и четкие, некоторые рефлексы вообще не заметны, они сливаются с другими, о чем это говорит? О том что вещество не имеет совершенной решетки и она как бы "кривая/косая"? Или это значит что то другое?
Вещество я синтезировал сам.
4. В данных которые привел Демидович, указаны параметры ячейки 6,5280; 6,5280; 3,8530. А в данных с документа который мне прислали указаны параметры: 6,549542; 6,549542; 3,842951. Почему они разные? Одни реальные (которые из справочника) и относятся к совершенной кристаллической решетке, а другие это экспериментальные и рассчитаны для конкретного вещества?
Спасибо.
-
Ребята, большущее Вам спасибо. Завтра вечерочком я буду пробовать разбираться! Вернее уже сегодня))
-
Демидович, большое спасибо.
Я извиняюсь за глупый вопрос, но где смотреть данные? Я не могу увидеть параметры для сравнения.
У меня есть рентгенограмма, есть данные рефлексов. С чем их сравнивать? Я чего то не понимаю.
-
http://materials.springer.com/isp/crystallographic/docs/sd_1627449 Там многие данные закрыты, но в понедельник могу скриншоты скинуть, нужно?
Да, гидроксид лантана нужен, что бы проверить с тем что определил оператор. Буду Вам очень признателен.
-
Огромное спасибо за ответы!
Электроны здесь не причем, они только генереуют рентген.
d - межплоскостное расстояние.
Лямбда длина волны характеристического рентгеновского излучения.
Напряжение на рентгеновской трубке 40 кв при токе 15 ма. Что бы знать длину волны, надо знать точно материал анода.
Точнее вы свободно можете выяснить в интернете, там подробно описываются и физические принципы метода вместе с математическим аппаратом и характеристические длины волн. Вообще же, это непростое дело, расшифровка рентгеновских лауэграмм.
Прошу прощения, я ошибся, не электроны там летят, а рентгеновское излучение, которое электроны выбивают.
Согласен что в интернете много справочников, но который раз убеждаюсь, что проще и эффективнее спросить у знающих людей, а уже дальше до разобраться самостоятельно, чем все с нуля разбирать.
На длину волны рентгеновского излучения, которая подставляется в формулу выше, если речь идёт о РФА. Думаю, что стандартным считается медный анод, но это нужно обязательно уточнить и посмотреть в справочнике соответствующую лямбду.
Порядок отражения - рентгеновский монохроматический луч может несколько атомных плоскостей пролететь и отразиться от каждой. Но как правило на этом не заморачиваются и подставляют единицу.
Спасибо за расшифровку буквы n.
Я про анод написал исходя из того что читал в интернете. Даже не знаю какой прибор стоит в лаборатории где мне делали анализ. Знаю что японский)).
В файле который мне прислали есть указанные рефлексы и табличка где все уже переведено. Я прикрепляю этот файл к сообщению. Это РФА анализ гидроксида лантана. (файл формата ворл.)
Исходя из формул, которую указали выше я сделал перерасчет и получил длину волны рентгеновского излучения.
Расчет:
В файле который прикрепил на странице 5 указаны углы в тета и там же дано межплоскостное расстояние.
Поставляя в формулу получаю следующее значение длины волны:
λ=2*d*sin Θ = 2*5.665*sin (15.63/2)=1.54059 нм.
Такое число получается при каждом вычислении. То есть длина волны равна 1,54 нм.
Оператор определил мне вещество. Но я хочу сам его определить. Я не могу понять где данные рефлексов гидроксида лантана? Они в файле который прикрепил не указываются? Там только указан номер карточки pdf? Эта база pdf я так понимаю платная. А существуют ли бесплатные базы, где можно посмотреть карточки?
Я вот нашел сайт: САЙТ, но там нет нужной карточки.
(Помню я уже задавал такой вопрос на форуме. Попробую поискать.)
Если есть данные справочника, то структура, чаще всего, известна. А при известной структуре дифрактограмму построить дело пары кликов.
В программе строить надо? Наверное это PowderCell? Я уже пробовал её освоить, но так и не смог.
Файл прикрепил к сообщению.
Ссылка на файл (РФА анализ гидроксида лантана): Ссылка (файл размещен на яндекс диске).
-
Большое спасибо за ответы!
А вы ничего не путаете? deg - единица измерения градусов, ангстрем - ед. измерения расстояний. В ангстремах часто измеряется длина волны рентгеновского излучения.
Скорее всего имеется ввиду зависимость длины волны и угла отклонения
Большое Вам спасибо. Думаю это то что нужно!
Конечно же я что то путаю)) так как понятия не имею о чем пишу
. Все в отпусках, спросить не у кого.Подскажите по формуле. d это то что нужно найти.
λ это длина волны электрона который летит на кристалл и отклоняется. Это должно быть указано в приборе или зависит от материала анода. В файле с анализом, это не указано, что странно, там только указано что 40 кВ и 15 мА току, и еще детектор SC-70.
А что обозначает буква n в формуле?
Осталось узнать из чего был сделан анод в рентгеновской трубке.
Думаю он там стандартный либо молибденовый либо вольфрамовый. А на что это влияет, подскажите пожалуйста? На длину волны электрона?
-
Уважаемые специалисты, объясните пожалуйста.
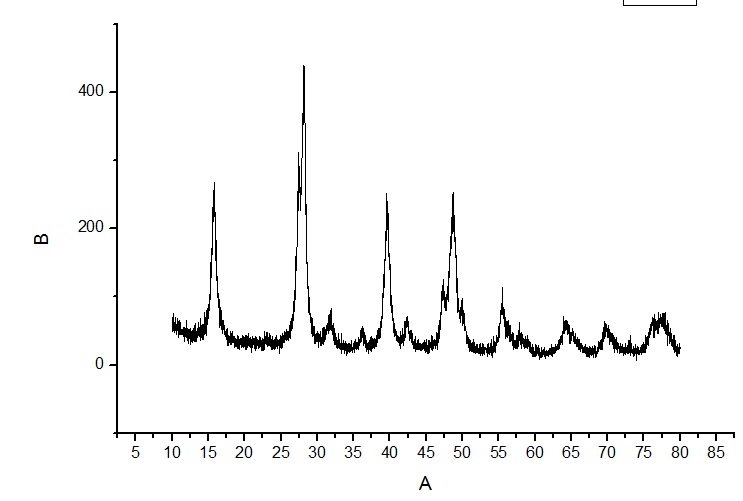
У меня есть данные РФА, график с рефлексами. Углы на графике показаны в 2Θ (deg). Но в справочнике для определения вещества углы указаны в ангстремах. Может это даже не углы, а что то другое.
Подскажите как перевести из 2Θ в ангстремы (d ang.), а то я не могу идентифицировать тогда вещества.
-
Маск, мне кажется что надо смотреть в области технологии шелкографии на стекле.
По температуре не сходится конечно, в ролике всего 100 градусов для закрепления изображения, в то время как в технологии выше 500. Но ролик может быть и уловкой. И эта наклейка светоотражающей подложки, это все может быть для отвлечения внимания и придания ноу-хау.
Почитать по технологии шелкографии на стекле можно тут:
-
Это лаба или НИР? Если вы планируете применять интегральные методы, то первым этапом, одну и ту же кривую (целиком) можно построить в линейных координатах для всех уравнений, и посмотреть на каком участке какое уравнение описывает эксперимент (где прямые).
Спасибо за ответ.
Это моя диссертация, руководитель сейчас занят каким-то отчетом, а я сам в другом городе и посоветоваться мне не с кем тут. По этой причине, я не могу сказать, какая проходит реакция.
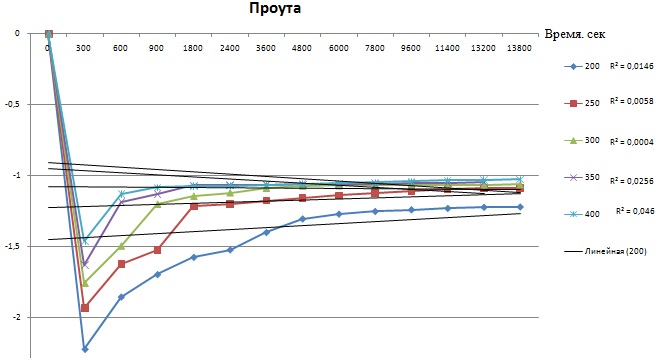
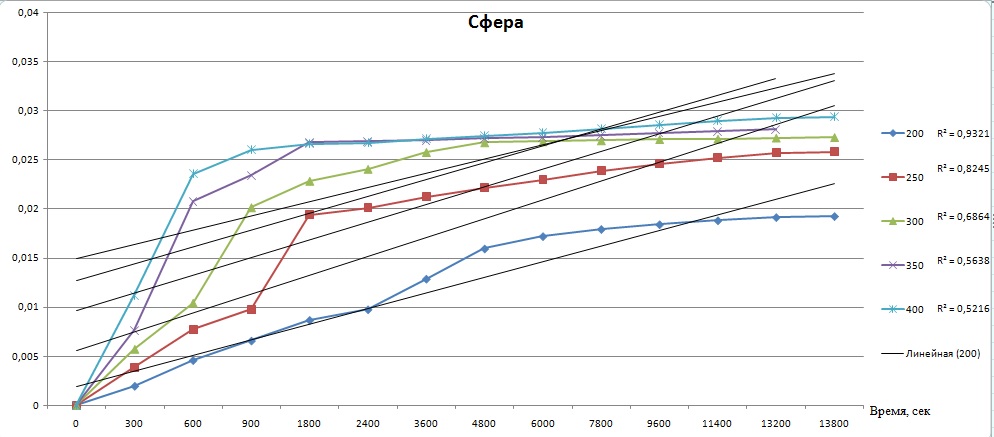
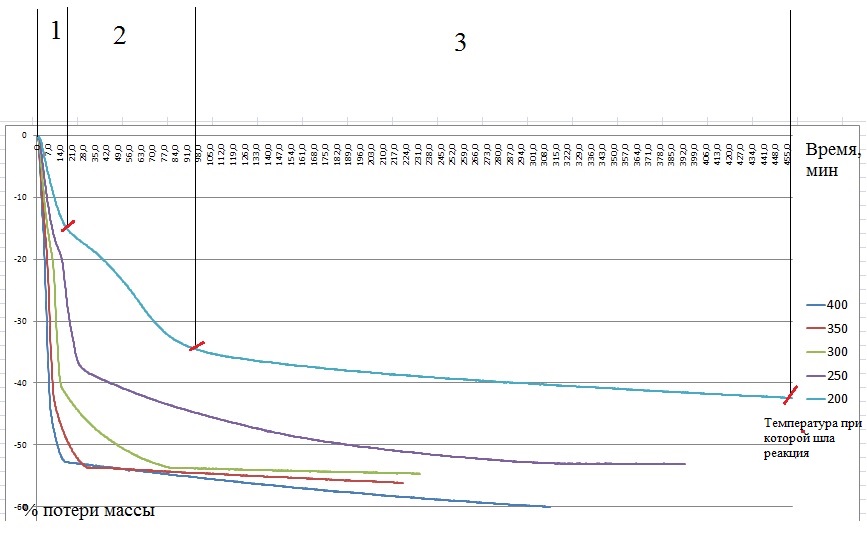
Я пробовал просчитать все кривые целиком. Графики ниже это результаты расчетов. Каждый график для каждого уравнения, они подписаны. В легенде, на против температуры, указана вероятность прямой. Я не вижу линеаризации, она вроде как происходит на кривой при 200 и 250 градусов и составляет больше 90%, но остальные кривые уже значительно ниже показывают эту вероятность. А по методикам необходимо 4-5 точек при разных температурах.
Вот сейчас до меня, кажется, начало доходить, что все кривые, вероятность линеаризации которых ниже 90% учитывать не стоит, и необходимо проводить эксперимент теперь только в интервале температур от 200 до 300 градусов и получить необходимое количество точек именно в этом интервале.
-
Здравствуйте.
Вопрос по расчету кинетики гетерогенной химической реакции. (Формулу химической реакции привести не могу).
Стоит задача, рассчитать энергию активации. Для этого надо провести серию экспериментов и построить графики потери массы от времени. График ниже.
Для расчета необходимо вывести формулу которая свяжет степень превращения вещества от времени и температуры. Используют следующие формулы: сокращающейся сферы, уравнение Яндера, Кранка-Гистлинга-Броунштейна и еще Проута-Томпкинса. Необходимо по этим уравнениям построить графики и определить по какому из них происходит построение прямой линии (с какой то вероятность конечно). Расчет ведется для одинакового интервала времени.
Вопрос: как можно произвести расчет, если реакция протекает в несколько стадий? На графике я обозначил эти стадии. И естественно с увеличением температуры эти стадии все менее заметны становятся, но все же их видно.
У меня в голове складывается ответ, что надо тупо разбить эти реакции на стадии и считать для каждой стадии кинетику по формулам. Но разве так можно делать, ведь время протекания реакции будет не одинаковым, как тогда быть?
-
Подскажите что может быть:
Стоит задача получить фосфат диспрозия.
Взяли хлористый диспрозий (DyCl3) залили его гидратом аммиака (NH4OH). Выпал осадок, гидроксид диспрозия и образовался хлорид аммония. Нагрели все это дело на плитке (300-400 градусов) что бы испарить хлорид аммония. Все высохло, остался только высушенный порошок.
Далее насыпали в порошок гидрофосфат аммония что бы получить фосфат диспрозия.
Прежде чем гидрофосфат аммония с диспрозием прокаливать при 1200 градусах надо испарить всю воду, по этому снова поставили на плитку и начали испарять влагу. Но что то пошло не так, сначала где то на 200-250 градусах образовалась какая то белая паутина, как сахарная вата, такие нити, тонкие тонкие и прямо срываются в вытяжном шкафу и уносятся. Подняли температуру еще, все нити испарились, но все что было в чашке стало черным, не прям черным но почернело. Что за ерунда, но это явно не то что то.
Та же самая процедура была проделана с тулием и в конце образовалось тоже самое, паутина и чернота. Что за ерунда может быть?
Реакции:
1 DyCl3+3NH4OH => Dy(OH)3+3NH4Cl
2 Dy(OH)3 + (HN4)2HPO4 => DyPO4......
-
Здравствуйте.
Алексей, спасибо понятно что все надо вбивать. Но к сожалению я совершенно не понял как и откуда брат данные. Пространственную решетку, вот откуда на берется, я так и не понял? Чувствую что это все бесполезно для меня, тут надо конкретно разговаривать со специалистом, а так я сам фиг разберусь.
Буду строить через эксель.
Алексей, скажите при отправлении статьи в журнал что надо обязательно показать на рентгенограмме, какие параметры? И, если представлять презентацию на докладе, в слайды вставить рентгенограмму - что там отобразить что бы было понятно что я действительно получил то вещество которое хотел. Вообще у меня возникают сомнения по необходимости представления рентгенограммы на докладе в презентации, кто там что разберет, это же надо быть специалистом и надо время что бы рассмотреть и проанализировать спектры. Подскажите как это обычно делается. спасибо.
Chaus, спасибо за базы. Правда я так и не смог понять как там найти гидроксид кальция.
-
Здравствуйте Алексей.
Большое спасибо, все получилось. Как же я сам не догадался воспользоваться блокнотом, это же ведь программа которая читает все подряд.
Все делаю как Вы сказали и теперь все открывается.
Кстати там по мимо файла с расширением .ras есть еще .DAT, и в этом файле в принципе тоже самое, те же числа для постройки графика и ни каких других данных нет. Поменяв расширение на x_y, его тоже можно считать для постройки графика в программе. Единственный момент, если сразу открыть его в программе, то он не откроется и программа вообще больше не включается, только переустановка. Надо через блокнот "зайти в файл" и в самом конце списка цифр удалить три энтера, после этого все запустится.
Алексей, снова прошу у Вас помощььььи :-).
С Вашей помощььььью разобрался как и где взять данные.
Но я не могу понять что делать дальше. Как теперь построить пики?
Загружая пример который Вы выложили в посте №12, есть окно с данными, прикрепил его к письму. Эти данные надо заносить самому при создании нового файла? Или как что делать?
Теперь данные есть, можно построить график и в Excel.
Конечно это не то что в программке специализированной, но можно...
Но лучше разобраться в программке.
-
Данные лежат в файле 2.ras. Вытянул, сделал файл 2.x_y. В принципе считает, но дифференциальная получается страшненькая, лучше не включать.
2.pdf - это тоже rar.Спасибо Алексей. Но как Вы это сделали?
У меня не получается извлечь данные. Я переименовал файл с расширением которое Вы указали, но открыть его не могу. При этом Ваш открывается.
Вы не могли бы помочь разобраться как работать в программе, мне просто надо делать и другие рентгенограммы, а это надо самому уметь делать.
Попробую поискать инструкцию, может кто писал как пользоваться, а так же методом тыка по изучать файл который создали Вы. Но пока я не понимаю совершенно как Вы это все строите.

 )) и пытался грузануть файл (поменяв его расширение в винраре).
)) и пытался грузануть файл (поменяв его расширение в винраре). 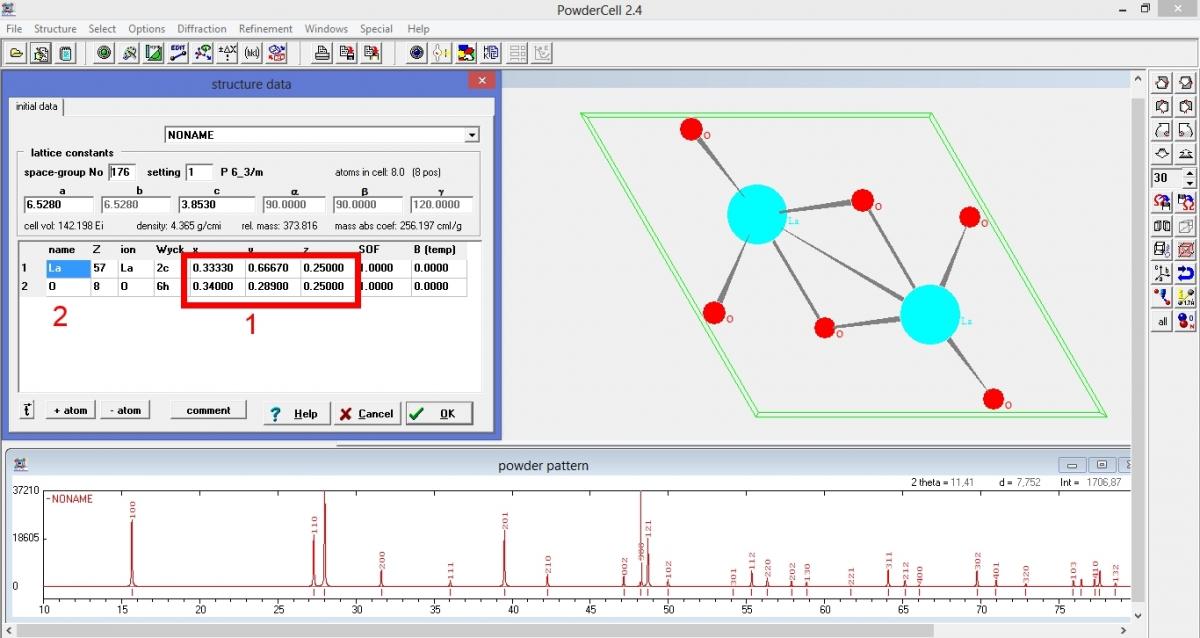
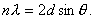

Программа Origin
в Общий
Опубликовано
Да, эту функцию я разобрался как использовать, но я хотел что бы эти значения, которые высвечиваются теперь в табличке, сразу же отображались на графике. Вот в чем дело, мне не точки надо посмотреть, а подписать данные. В любом случае, спасибо.
Сделал подписи в ручную в экселе.
Программу которую Вы указали посмотрю, спасибо.