igorchem
-
Постов
91 -
Зарегистрирован
-
Посещение
-
Победитель дней
1
Тип контента
Профили
Форумы
События
Весь контент igorchem
-
если государство поднимет налог на энергоносители и один киловатт/час будет стоить 30 евроцентов (как в Европе сейчас), то очень будет окупаема.
-
ТС, а может проще все-таки турбу в ебее купить из США, там за полштуки довольно не убитые продаются, а управление к ней, Вы, как я понимаю, сами сколхозите - там на любом паршивеньком микроконороллере на раз все делается. У турбы обычно стандартный трехфазник, правда с большим диапазоном, то, что я видел и пользовал - стартует с 30 об/мин и 0.6В (не помню сколько миллиампер), а при работе выходит на 84000 Об/мин и 19В / 6А, кажется это был какой-то из ерликонов, но и у эдвардсев все примерно также.
-
есть тиратроны, которые на 50КВольт и выше работают, я даже когда-то в предыдущем своем проекте такое пользовал. Рентген с них валит неподетццки, хотя внутри ничего радиоактивного нет. Использовать в лампах радиоактивные геттеры - себе дороже - они будут не предсказуемо ионизировать остатки газа внутри лампы и у лампы будет левая вольт-амперная характеристика, поэтому так никто не делал и не делает. В качестве геттеры проще всего взять сплав V-Zr или Ti-V-Zr или мелкодисперсные порошки в несколько микрон из оных металлов примерно в 1:1 или 1:1:1 мольном соотношении. Если объем геттеры примерно равен одной 500-ой всего дегазируемого объема, то после форвакуума можно на 1е-7 Торр выйти, сам проверял. Но нужно гарантировать, что внутри больше ничего газить не будет, а это - ой как сложно. Геттеры надо перед использованием прогреть на 500С или чуть выше на 1е-2 Торра - ну типа в вакууме от форвакуумника. Сами порошки дешевле всего заказать у китайцев, если рядом ни у кого не найдете.
-
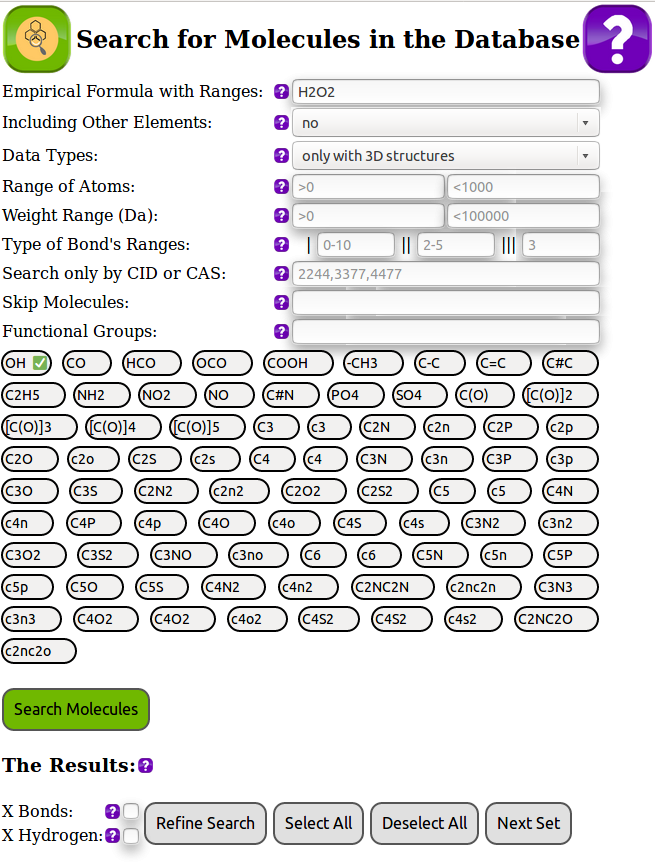
Спасибо большое за отзыв! Согласен! На данный момент уже запланировано: 1. прикрутить WebGL и рисовать 3Д в виде объемных шариков, или шариков и палочек (типа chemcraft но с тенями и перспективой), вне браузера на OpenGL это уже работает, но пока в базу не встроили. 2. переключать несколько молекул (стереоизомеров и изотопных изомеров) на одной картинке, 3. опционально переключать очень схожие молекулы внутри одной картинки, как сейчас реализовано переключение конформеров. Например один и тот же лиганд + все катионы, сейчас оно все валится в разные картинки, а по сути там обычно схожие конформеры и их идеологически правильно смотреть вместе друг с другом. Сразу каждую молекулу в отдельный фрейм выводить не хотелось бы, хотя конечно для каждой найденной молекулы можно опционально выводить ее отдельно. Да, обязательно! Эти величины внутри базы конечно имеются, но они просто еще не попали на экран Мы машинным обучением в мол-динамике тоже грешим - это одна из целей построения такой базы. верно, допиливаем, туториал обязательно сделаем, зачатки его уже имеются, если нажать кнопку с вопросом в верхнем правом углу
-
Ой, только сейчас заметил, что у нас по-разному сортируется набор молекул во время отображения. Зафиксил, чтобы всегда было одинаково. Сейчас при поиске выше сорбоза в списке на 8-м месте. Есть довольно большая проблема с аналогичными названиями, сорбоза в нашей базе фигурирует по ее ИЮПАКовскому названию, то есть как 1,3,4,5,6-pentahydroxyhexan-2-one
-
Спасибо большое, chemister2010 за отзыв! С названиями и синонимами - сложно, будем думать, будем ли мы это делать или нет, у нас названия запрятаны в самом конце индексации и, часто названия из PubChem какие-то очень не адекватные, поэтому мы на них индексацию забили. спасибо, да, это легко исправить, на днях сделаем! Синонимов - в смысле схожих молекул? Если да, то тут надо по-другому поступать. Надо выбрать чекбокс под молекулой, для которой Вы хотите найти аналог, и подняться выше и выбрать Refine Search, попопутно выбрав удаление водородов или игнорирование типов связей. Также мы сейчас перестраиваем базу, чтобы на одну картинку попадали все стереоизомеры и все изотопные изомеры. Иначе иногда даже изотопных изомеров в нашей базе оказывается под сотню и их очень не удобно все пролистывать. На днях будет... сейчас на это все индексация строится, просто это неделю по времени занимает. Странно, только что проверил - пятая структура. Должно находиться, вот как у меня выглядел поиск перекиси водорода: и результат был вторым в таком поиске. Углы (95.91 и 95.60) слегка не симметричны в пределах расчетной погрешности, только что проверил - там до градуса изменение этого угла дает меньше изменение общей энергии, чем изменение на 0.1 градуса по торсионному углу. Точность сходимости у нас около 0.005-0.01Ангстрем по координатам, ибо точнее уже без очень недетских ab initio не вытянуть.

-
Встроили отображение расстояний между атомами, обычные и торсионные углы. Также добавили набор радикалов (предварительный). Рассмотрим пример, допустим мы хотим посмотреть сорбозу, у нее формула C6H12O6, записываем ее в поле Empirical Formula with Ranges, вспоминаем, что в ней есть C=O группы и -[CH(OH)]4-, отмечаем ниже поля CO и [C(O)]4 для этих функциональных групп в формате SMARTS - SMILES и получаем довольно небольшой набор молекул, которые подходят под критерий нашего поиска. На 5-ом месте как раз расположилась наша искомая сорбоза. Кликаем на 2D -> Conformer#1 и маркируем двойными щелчками последовательно до 4-х атомов. Внизу под молекулой отображается длина связей, углы и торсионные углы. При переходе от одного к другому конформеру эти величины меняются в соответствии с номером конформера.
-
да не буду спорить, пусть будет по Вашему.
-
известны и позволяют сделать ppb по пространству и времени (ppb/s) - это две большие разницы
-
Вангую, что моль с Дальтоном перепутали
-
так в пульсе для ЯМРа сложно использовать - отклик идет долго, частенько дольше секунды, а стабильность магнитного поля по пространству и времени должна быть единицы ppb
-
не, не вижу смысла - это - к Брукеру или Жеолу. 7 Тесел на сборных неодимовых магнитах еще 20 лет назад делали, но было не технологично, на нашем материале теоретически где-то 8-10 тесел сделать можно, просто мы еще не отмасштабировали наше производство до фокусированных сфер около метрового диаметра. Ну а когда получится - оно же совсем энергию потреблять не будет. Наши 200Ватт потребления - это где-то 50 Ватт на шимминг магнитов + питание электроники самого ЯМРа и динамической ядерной поляризации, включая питание несколько сотен процессоров для обработки многомерных спектров с неравномерным семплингом. Получаемые 7-20- мерные ЯМР спектры, еще в добавок сильно разреженные, трактовать-то простой человек не может, вот и приходится изгаляться, и с помощью квантовой механики эту задачу решать. Про такие разреженные спектры (правда не на нашей железке) - doi: 10.1038/nmeth900 - еще 15 лет назад все было понятно, но вот удобная железка для этого - да, существенно подзадержалась и есть надежда в ближайшее время исправить эту ситуацию.
-
Да, так конечно можно, но мегаватты-то жалко. Хочется чего-то удобного, чтоб у каждого в лабе стояло и в прямом и переносном смысле. У нас пока концепт где-то до 4Теслы в бенчтопе размера с системный блок компьютера и весом где-то 15кг с потреблением около 200 ватт и входом к форвакуумному насосу (не турбомолекулярке) для поддержания всего, что связано с динамической ядерной поляризацией. Чувствительность по спектрам при этом получается примерно как на криогенной 300ке.
-
Не, к сожалению, не работал. В жидком азоте вариантов существенно больше, но как раз не хочется, так как это сильно удорожает аппаратуру (мы ЯМРы с ядерной поляризацией делаем). У нас цель в магнитах 10 Тесел без криогена, пусть большой байдой хоть метр в диаметре, но обязательно без криогена. Сейчас как бонус - из самых дорогих компонент только висмут, все остальное кобальт, марганец и железо понятно цену не определяют.
-
после 2.4 Тесел где-то даже пермендюр как парамагнетик себя ведет, а стальной шарик и того раньше, то есть главное быстро им не дергать, чтобы токи Фуко не создать. Вы попробуйте толстый кусок алюминия из 13-ти теслового ЯМРного магнита быстренько высунуть, он реально как камень встает. а как магнитная жидкость плотность повышает? За счет какого эффекта?
-
Спасибо за интерес! Да, постоянные фокусированные магниты. Вроде как при современном известном сопротивлении меди и серебра без физического отвода тепла вроде как электромагнитные не сделаешь, а вот постоянные - да, можно даже без редких земель сделать, правда с фокусировкой: https://patents.google.com/patent/US10646722B2/en В принципе люди на неодимовых и выше достигали, у нас тоже есть надежда на 10 тесел, но пока в небольших габаритах (10 см внешние размеры) и с приемлемой для ЯМР равномерностью поля, где-то не больше 4 Тесел и получается. Состав в 9-ом клейме.
-
Согласен с Вами, но, не для задачи ТС. Я как раз о том, что без реально хорошего коатинга все юзом пойдет. Мы варили магниты, чтобы достигать 4+ Тесел без криогена (номерок патента и ссылка на него у нас на сайте имеется), и получаемый у нас состав и конфигурация имеют хорошую проводимость. На заре этого исследования у нас было желание чем-то покрыть магнитные частицы, чтобы проводимости магнита не было, понятно, что стеариновые варианты рассматривали, но с поверхности частиц большинство покрытий просто сразу сдувается, ибо уже в одной тесле частицы очень крепко друг к другу начинают липнуть. Даже пробовали париленом покрыть, но это реально гемор, хотя он-то как раз держал. Правда его толщина получалась огромной, то есть треть магнита, и две третьи - парилена. ИМХО, задача ТС по физическим принципам схожа с тем, что мы делаем, когда намагничиваем магниты из нашего магнитного материала. В результате мы от покрытия отказались, и стали плавить магниты, сохраняя проводимость, а вот ТС это, к сожалению, не подойдет. Из этого всего я высказываю свое робкое ИМХО, что без покрытия или без перехода на токо-не-проводящие частицы, ТС свою задачу не решит.
-
может лучше цитрат взять? Но, как выше уже заметили, железо и никель - проводники, и все юзом у ТС пойдет. То есть тут при использовании пирофорного железа основная проблема не в том, чтоб его получить, а в том, чтобы его закоатить - то есть покрыть чем-то непроводящим. У нас была недавно задача при производстве наших магнитов на проводящем материале, было тоже желание покрыть частицы магнита чем-то. Из подходящего получилось, что подошло покрытие из поли-пара-ксилилена, но реально геморно. Может все-таки сразу взять что-то непроводящее?
-
Я бы на месте автора таки не смотрел бы на проводящие вещества с высоким мю и не изобретая сердечник с порошковым железом, а взял бы c90 сердечник веррокскубовый и раздолбал бы его в ступе. При должной сноровке это легко осуществимо. По идее даже в спирте потом его можно будет гонять, хотя, ИМХО, безопаснее при долговременном пользовании - в неполярных растворителях, ибо в присадках там щелочноземельные элементы есть, и долговременно со спиртом они могут прореагировать.
-
Спасибо большое за отзыв! Да, действительно, у нас в базе для каждого конформера имеются координаты всех атомов и из них можно получить и длины связей и углы, включая торсионные. Пока эта возможность у нас есть только во внутреннем интерфейсе базы, но мы как раз собирались как-то выдавать эту информацию пользователю по его желанию. Под каждой найденной молекулой есть возможность выбрать 2D и список (если есть) Conformer #1,.... Conformer #10. 2D - это обычный depict с PubChem - только для удобного отображения плоского рисунка, а вот в Conformer #1,.... Conformer #10 все конформеры с 3D координатами. Эти конформеры сейчас на экране можно вращать мышкой и смотреть на них с разных сторон. Мы планировали выдавать для каждого конформера и углы, и длины связей, и координаты, но пока не придумали как было бы это удобно делать. Если бы Вам было бы не сложно посоветовать, как это удобно было бы эту информацию видеть, с радостью прислушаемся и, надеюсь, скоро добавим! Сам вижу, что, например, по двойному клику на атомы мы маркируем от двух до четырех атомов, и в зависимости от того, как они связаны, выдаем Ван-дер-Ваальсовы расстояния, обычные расстояния, углы или торсионные углы.
-
Ссылки на наши стати и патенты лежат у нас в открытом доступе на нашем сайте, а Вы, как всегда, поленились посмотреть. не увидел в Ваших постах ни единого аргумента чем она Вам не понравилась, только истерика с Вашей стороны, с призывами, чтоб мы начали учиться устанавливать какое-то легаси 90-х годов. Чтобы иметь право наставлять, Вам, уважаемый химхлам, нужно быть видным ученым с мировым именем, поэтому мне очень интересно узнать Ваш ORCID, чтоб оценить Ваш вклад в мировую науку и после этого принимать решение, стоит ли прислушиваться к Вашим советам, или все-таки отнестись к Вашим советам со здравым скептицизмом. Очень жалко, что Вы не идете в ногу с мировой наукой, размер базы данных PubChem - около 40 Терабайт, наверное Национальной Библиотеке Медицины такие базы зачем-то нужны? Может все-таки, уважаемый химхлам и Вам настало время забыть legacy 90-х годов на 100 мегабайт и поддерживающее, как Вы сказали, максимум пол миллиона структур, у нас-то пол миллиарда структур уже сейчас и нашу базу покупать не надо - она бесплатная!
-
статьи, в том числе в Нейчере, да и и патенты у меня есть, удивительно, что Вы их еще не нашли, мой хирш пока только 11, а у Вас? А на форуме есть живое общение, в том числе с теми, кто может и в штыки воспринимает, как Вы, например - или Вы тоже из тех, как Вы назвали, школьников и двоечников? Надеюсь, что нет, поэтому, надеюсь, что не зря распинаюсь ))) А Вы точно уверенны, что без суперкомпьютерной версии IsisBase сможет проиндексировать пол миллиарда конформеров? Вы хоть понимаете, сколько это информации? Пол миллиарда конформеров - при, в среднем, 50 атомах, и 3-х декартовых координатах - это уже 75 миллиардов чисел (в двойной точности это 600 Гигабайт), а к этому надо добавить еще много информации по структуре, типу атомов, которая еще примерно столько же занимает и про индексацию не забывайте, которая в большинстве баз еще удваивает размер. Я вот сомневаюсь, в тех рекламах, что я находил об IsisBase, как-то даже про миллион молекул не говорят. Вот Вы про нее много говорили, значит, или она у Вас есть, или часто пользовались. Сколько конкретно молекул и конформеров у Вас там было, давайте честно сравниваться? Вы, кстати, наш поиск хоть раз попробовали или Вас тоже Роспотребнадзор не пускает? Так если не пробовали, зачем комментировать? А так бы повторили бы пример, что я написал выше, или придумали бы сами что-то для Вас интересное и выложили бы здесь свой опыт по использованию на всеобщее обсуждение!
-
Все люди в нашей команде имеют химическое образование, я дополнительно еще математик. Многие из нашей команды кандидаты и доктора наук с большим опытом работы. Вас, я вижу, сильно напрягло, что база делается для людей - я мечтал еще лет 30 назад, будучи студентом, иметь такую под рукой, и база будет распространяться бесплатно. Ведь не каждый химик может позволить себе купить базы поиска. Сейчас мы дорабатываем и пополняем программу. По откликам пользователей сделаем все, чтобы она работала хорошо, и люди были довольны. Я понимаю Ваше настроение: многие новое и прорывное воспринимают всегда в штыки - это же классика жанра. Каждый человек, хоть как-то связанный с органикой, понимает на сколько важно иметь точное понимание пространственной структуры молекул, которые участвуют в синтезе, чтобы избежать стерических затруднений в реакциях или не сварить рацемат вместо хирально чистого вещества. Наша база позволяет сразу взглянуть на все молекулы, которые органик планирует синтезировать. Рассмотрим простой университетский пример. Мы хотим сварить хлорпроизводные кофеина, также хотелось бы иметь под рукой список таких хлорпроизводных с их пространственными структурами, плюс хотелось бы иметь ссылки на патенты и другую литературу об этом процессе. Итак, во вспомогательном поиске у нас в базе вводим C8H10N4O2 и долистываем до кофеина. После этого, выбираем X Hydrogen, чтобы игнорировать водороды в следующем поиске. В этом случае, в строке Functional groups, у нас имеется магическая запись 2519H - чтобы был остов кофеина без водородов. Далее выбираем в основном поиске C8-10 N4 Cl1-5 - то есть мы к остову кофеина без водородов до двух дополнительных атомов углерода готовы посмотреть, а также от одного до пяти атомов хлора мы тоже хотим увидеть. Смело жмем на поиск и получаем набор известных на данный момент хлорпроизводных. Для каждого из которых мы можем выбрать (если есть в базе Pubchem) конформер и посмотреть на него, покрутить его в 3Д. Почитать по ссылке на PubChem кто и когда это патентовал, кто производит, кто продает. К сожалению, Вы сильно ошиблись, недооценив сложность алгоритмов поиска. Если бы мы, как Вы смешно предположили, считали "на счетах", то Вам бы пришлось комбинаторным образом перебирать все структуры и считать конформеры, пусть с помощью Вашей бесплатной программы. При расчете Вы могли забыть что-то или просто не дождаться, пока Вы переберете сотни и тысячи этих конформеров. Они могут быстро считаться, но хотя бы минуту расчетов на простую молекулу без flexible bonds надо потратить, а обычно час на конформер может потребоваться, если у Вас в кустах нет суперкомпьютера. В мною указанном выше примере перебирается больше тысячи таких конформеров! Органик не обязан знать все параметры настроек программ по квантовой механике и, если он сам, без помощи квантовика, это начнет делать, может лажа получиться. Мы считаем, что наш удобный поиск - это будущее в органической химии, и с радостью учтем все советы по существу, что еще может дополнительно в поиске и в отображении результатов Вам быть интересно!
-
Странно, что Вы с этого не начали, а вспоминали MDL. То, что я на этом форуме здесь на всеобщее обозрение предоставил, не означает, что там, где я работаю, ресурсы по вычислительным алгоритмам недостаточные, чтобы создать адекватную поисковую систему. Вас, кстати, это не интересовало, как собственно и алгоритмы поиска, которые мы используем. Не услышал от Вас адекватных вопросов в этом направлении. То есть, из всей дискуссии с Вами, я вижу только одно - Вас почему-то очень покоробило и возмутило, что кто-то "новенький" на форуме предложил бесплатный продукт, который позволяет искать по огромной известной в мире базе (на данный момент это PubChem с пол-миллиарда конформеров + то, что мы уже успели домоделировать), и Вы, после этого, просто не разбираясь, решили высказать свое необоснованное фи, что де никто, кроме Вами сейчас известных фирм (которых Вы даже не сразу вспомнили) не может такое, и, вдобавок, бесплатно предложить. Давайте с Вами спор закончим ))) Вы даже сразу названия фирм в этой области сказать не можете, а темы о патентах и алгоритмах Вы намеренно игнорируете, очевидно, Вы не совсем хорошо в этих темах разбираетесь.
-
MDL исчезла с рынка уже 7 лет назад, их сайт mdl.com сайт давно захвачен доменными перепродавцами, скорей всего это означает, что фирма банкротилась, а не продавалась. Фирмы уходят, приходят на их место другие, это нормальная конкуренция. Раньше на счетах считали, а теперь используем суперкомпьютинг и заменяем целый НИИ. Для того чтобы дискутировать, надо никогда не недооценивать соперника. Про наш велосипед Вы мало чего знаете, Вы услышали только звонок от велосипеда. Поэтому дискутировать дальше бессмысленно. Люди пользуются программой и говорят спасибо. Тем более, что пользование бесплатно.