mariapetrova38445
-
Постов
4 -
Зарегистрирован
-
Посещение
Тип контента
Профили
Форумы
События
Весь контент mariapetrova38445
-
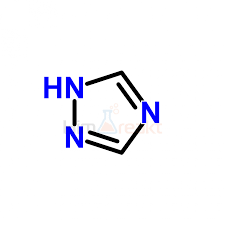
Здравствуйте! Подскажите, пожалуйста, в молекуле 1,2,4-триазола четыре связи C-N являются ароматическими??

-
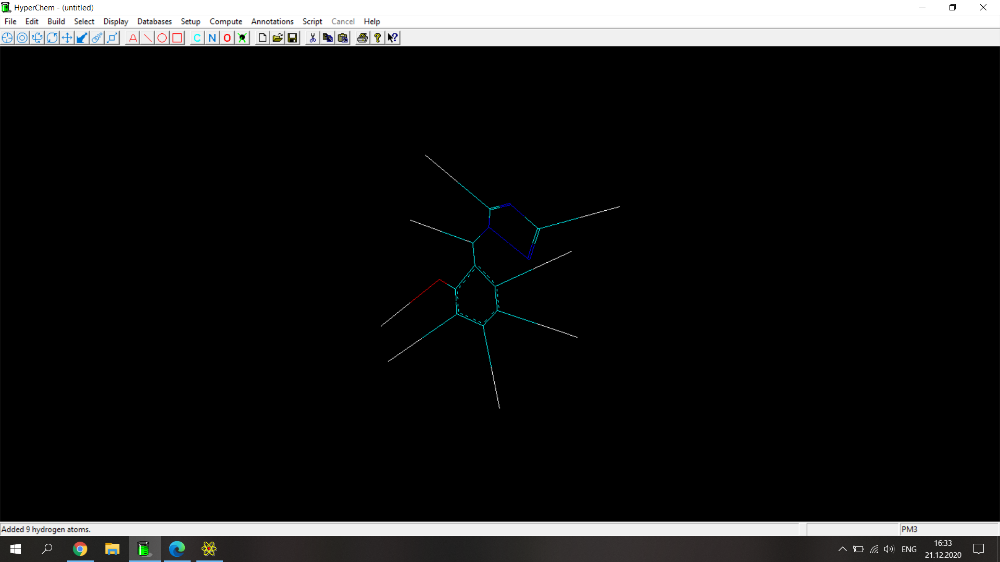
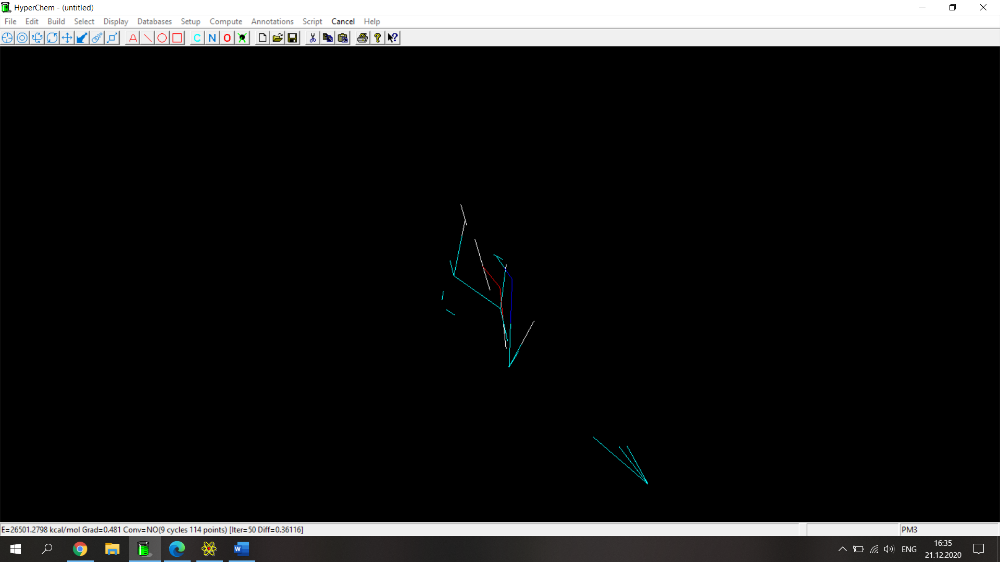
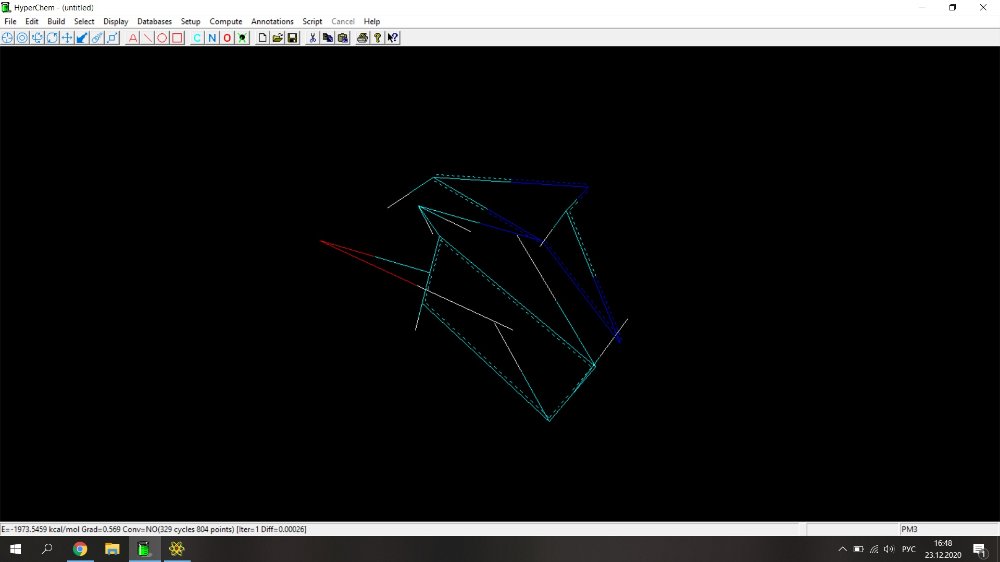
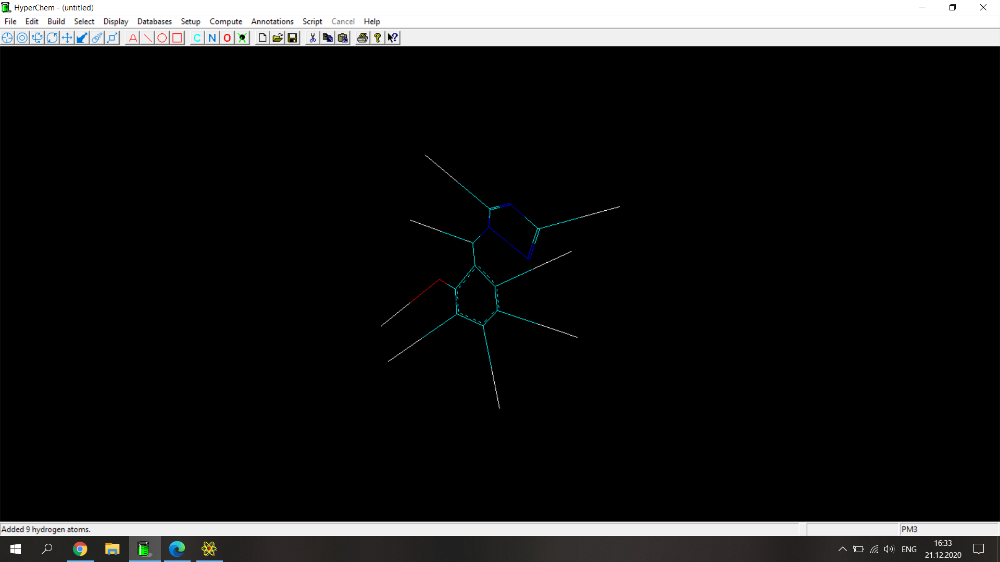
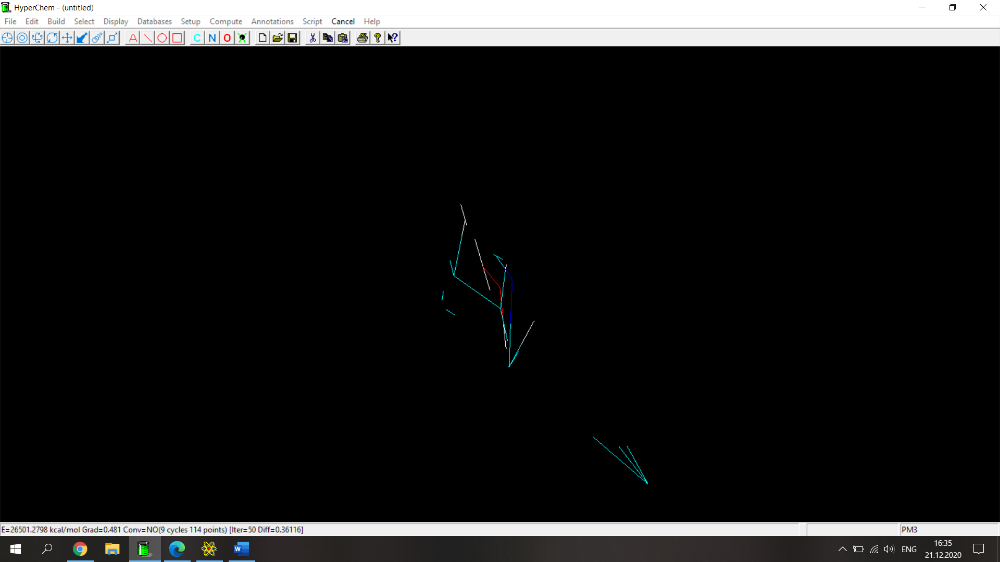
Здравствуйте! Возникла проблема при расчете физико-химических параметров (дипольный момент, поляризуемость, площадь поверхности, площадь поверхности на плоскость, липофильность, объем и энергия сольватации) с помощью программы HyperChem. Действую по следующему алгоритму: рисую молекулу, нажимаю Build->Add Hydrogens. Затем Setup->Semi-empirical->PM3. Потом Compute-> Geometry Optimization-> Polak-Ribiere (0.1 kcal/mol и 300 maximum cycles). Прикрепила скрин с молекулой 2-(1Н-1,2,4-триазол-1-илметил)фенола (скрин 1), расчет идет не до конца, не пишет внизу "YES" и вид молекулы после оптимизации очень смущает (разрозренные по всему экрану связи)(скрин 2), либо просто не досчитывает до конца (скрин 3). Поэтому невозможно перейти по вкладке Properties и найти дипольный момент, а во вкладке QSAR properties приведены данные, которые очень сильно отличаются от данных, полученных для похожих веществ.
-
В водных растворах фосфорная кислота диссоциирует ступенчато. Диссоциация на каждой следующей ступени протекает слабее, чем на предыдущей. Полностью на ионы фосфорная кислота не распадается и относится к кислотам средней силы
- 1 ответ
-
- 1
-

-
Здравствуйте! Возникла проблема при расчете физико-химических параметров (дипольный момент, поляризуемость, площадь поверхности, площадь поверхности на плоскость, липофильность, объем и энергия сольватации) с помощью программы HyperChem. Действую по следующему алгоритму: рисую молекулу, нажимаю Build->Add Hydrogens. Затем Setup->Semi-empirical->PM3. Потом Compute-> Geometry Optimization-> Polak-Ribiere (0.1 kcal/mol и 300 maximum cycles). Прикрепила скрин с молекулой 2-(1Н-1,2,4-триазол-1-илметил)фенола, расчет идет не до конца, не пишет внизу "YES" и вид молекулы после оптимизации очень смущает (разрозренные по всему экрану связи). Поэтому невозможно перейти по вкладке Properties и найти дипольный момент, а во вкладке QSAR properties приведены данные, которые очень сильно отличаются от данных, полученных для похожих веществ.