Alex021
-
Постов
16 -
Зарегистрирован
-
Посещение
Достижения Alex021
")





-
Этот вариант не герметичен, HCl будет бесконтрольно диссоциировать в воздух лаборатории. В герметичном варианте можно точно рассчитать сколько мне нужно H2SO4, NaCl и H2O чтобы получить нужную концентрацию с минимальными потерями HCl.
-
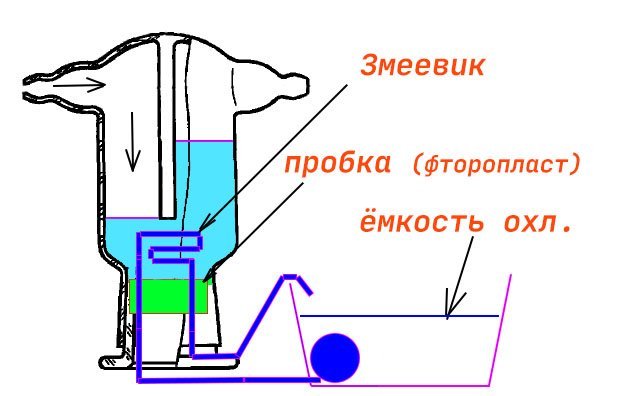
Самый лучший способ избежать засасывания кислоты - использовать склянку Тищенко. Это даст герметичность. Но пока не нашел где купить по вменяемой цене... И еще возник вопрос - как остужать кислоту в склянке? для концентрации 36% при температуре +42°C изменение энтальпии ΔH=-140.6, то есть нагрев дистилл.воды составит 140 °С. Думал подавать сухой лёд (CO2) но в интернетах пишут что существует такая реакция 2HCl + 2CO2 → 2HCO2 + Cl2↑ и это всё испортит. Поэтому выбор остановил на скл.Тищенко для сыпучих поглотителей. У неё внизу широкий шлиф. Думаю сделать пробку из фторопласта, а на ней сделать змеевик из тонкой стеклянной трубки. В процессе повышения концентрации HCl в скл.Тищенко через этот змеевик пропускать воду, которую в соседней ёмкости буду остужать сухим льдом. Такой самопальный холодильник. Правильно ля я мыслю?

-
для настоящих химиков это наверно как 2х2, а мне все это в новинку. Парциальное давление только-только начал понимать. По-ходу пьесы делаю всякие расчетные таблицы на Excel'e.
-
Похоже так и есть. Думаю чем наполнить сосуды перед реакцией (заменить воздух)? Имеющиеся сварочные Ar и CO2 имеют молярную массу больше чем HCl. В реакторе хорошо - больше HCl выйдет в Вюрца. А вот в приемнике плохо - эти газы осядут вниз и не дадут контактировать HCl с поверхностью воды. Может посоветуете какой-нибудь безводный газ, который несложно получить, чтобы он был тяжелее воздуха, легче HCl и безопасный для здоровья ? Идея создать газовую "прокладку" в колбе-приёмнике. Если HCl перенасытит раствор и пойдет через верх, то влажная лакмусовая бумажка в верху колбы-приёмника маякнёт что уже перебор с концентрацией. Или это я излишне загоняюсь и лучше рассчитать и отслеживать объем/вес жидкости в колбе-приемнике?
-
Да, все верно. Я о том же. Подскажите какую литературу надо читать чтобы освоить этот вопрос? Гугл и Яндекс ничего толкового не дали. Имею в виду именно про скорость поглощения в зависимости от площади контакта, температуры воды и газа, парциального давления и других факторов. Чтобы можно было на Excel-калькуляторе рассчитать диаметр и глубину воронки, скорость подачи H2SO4 в NaCl.
-
Если начнется процесс засасывания, тогда можно приоткрыть пробку "В" у капельной воронки - через компенсатор давление уравняется. И в этом случае HCl, который находится в трубке, будет еще растворяться в стакане "Е". Посмотрел ю-туб. Там даже барботажа нет и тогда действительно чем больше площадь соприкосновения с "водой" тем быстрее процесс растворения? Похоже я просто не понимаю как HCl растворяется в воде. Подскажите, где можно подробно почитать про скорость растворения хлороводорода в жидких растворах с различными концентрациями? При насыщении раствора скорость поглощения будет падать же? Надо бы рассчитать скорость генерации HCl и уравновесить её со скоростью поглощения. Тогда можно сделать с минимальными потерями HCl, а так же рассчитать эффект засасывания.
-
 Alex021 изменил фотографию своего профиля
Alex021 изменил фотографию своего профиля -
Нет уж. Надо пробовать. У меня если пуля в голову попала, то покоя она мне уже не даст, пока не сделаю.
-
Ну я делал простые неопасные растворы для изготовления печатных плат. Там инструкции - ребенок разберется. После этого говорить что я что-то химичил - язык не поворачивается. Захотелось похимичить посерьёзнее. Прикупил всяких стеклянных элементов - конструктор чтобы собирать различные аппараты. Теперь оказывается что не все так просто, много нюансов, да и рассчитывать надо уметь самому. Вспоминал что за "моль" была на уроках химии в школе. Ну и концентрированные кислоты - это уже опасненько. Если что-нибудь бабахнет, думаю это будет совсем не так весело как в Ералаше. Тут лучше 7 раз отмерять и еще 10 раз перемерять... Поэтому помощь настоящих специалистов на форуме для меня бесценна.
-
Спасибо, pauk. Очень важный нюанс. И тема то что надо! Засасывание происходит при остывании аппарата? Я совсем новичок в химии, но мне кажется что барботажем газ лучше растворится в жидкости, чем просто касаясь поверхности. Наверно полезно после Вюрца поставить холодильник Либиха или спиральный - в них много не засосет, и они будут дополнительно остужать хлороводород перед экзотермическим барботажем в скл.Дрекселя? Я планирую ставить 2 склянки Дрекселя последовательно. Во 2й будет раствор пищевой соды - это чтобы хлороводород не выходил в воздух из аппарата. Наверно на выход 2й склянки Дрекселя надо будет подключить вакуумный насос, а то давление из реактора может не продавить 2 барботажа и будет травить через шлифы. В любом случае перед началом работы любого аппарата я сначала на вход ставлю хлор-кальциевые трубки, а на выход вакуумный насос - чтобы вытянуть влажный воздух из аппарата и заменить его сухим. Могу наполнить аппарат Ar или CO2. Может излишне загоняюсь, но хочется получить продукт максимально приближенный к идеальному. Есть ещё мысли проводить реакции под вакуумом потому что температуры кипения растворов понижаются - реакции идут более интенсивно. Тогда точно не засосет в реактор. Только боюсь как бы не лопнул какой-нибудь стеклянный элемент аппарата.
-
Угу. Вот только КАК? Я вижу 2 этапа: 1 - выпарить азеотропную смесь 2 - с помощью серной кислоты отнимать воду у азеотропной смеси, таким образом высвобождая газообразный HCl, который потом промывать в склянке Дрекселя через другую порцию азеотропной смеси. 1й этап нужен чтобы поменьше расходовать серную кислоту на 2м этапе. FeCl3*6H2O имеется. Но нужно понимание как идет процесс. Как рассчитать пропорции? Какие условия прохождения реакции? Какой аппарат собирать для этого? Какие "грабли" где лежат?
-
Попробую сконцентрировать вопросы: Если температура кипения азеотропной смеси HCl*H2O при давлении 1атм. = +108.584°С, а температура кипения воды +100°С, тогда это близкокипящие жидкости. Как их разделить? Читал что для этого используется дефлегматор и опционально насадка Дина-Старка. Опыта у меня нет. Есть у меня дефлегматор 200мм на шлифах 14/23. Он подходит к насадке Дина-Старка. Но мне кажется что он совсем маленький и плохо справится с разделением. Думаю купить большой дефлегматор 400мм 29/32. Но под этот дефлегматор не могу найти насадку Дина-Старка. Колхозить эту насадку из разных элементов думаю не лучшая затея. Получается этот дефлегматор (400-29/32) ставить прямо на нагреваемую колбу. И следить за температурой на Вюрце чтобы не превышала +105°С. Так? Это будет выпариваться вода. Когда кипение прекратится, то заменить колбу-приёмник, обернуть дефлегматор фольгой, затем разогреть оставшуюся смесь до +120°С на Вюрце и получать в приемник чистую азеотропную смесь? А всякие присадки, которые не выкипели с водой, останутся в нагреваемой колбе. Где гарантия что они не будут выкипать вместе с азеотропной смесью? Как проверить чистоту полученной азеотропной смеси? Нужно ли после холодильника Либиха ставить ещё спиральный холодильник для лучшего остужения азеотропной смеси? Просто после выпаривания воды на стенках холодильников будут присутствовать остатки воды, а площадь спирального намного больше чем Либиха.
-
Я такого не знал. Спасибо. Сейчас, изучая хлористый нитрозил, встретил метод его получения нагревом смеси соляной и азотной кислот. HNO3+3HCl → Cl2↑ + 2H2O + NOCl То есть у меня нормативкой уже было заложено травление, например, высоконикелевых сплавов именно хлористым нитрозилом, только про него прямо ничего не сказали, а сказали травить в кипящей смеси HNO3+3HCl. Хотя с NaNO3 вроде бы выгоднее - нет кипения, нет Cl2 на выходе. Чем больше узнаю, тем интереснее становится химия.
-
Не сказать чтобы совсем не травит, но получается долго и некрасиво. Требуются смеси концентрированных кислот (HCl+H2SO4, HCl+HNO3). Для различных хим.составов различные смеси. Без воды (условно). В некоторых случаях темплет нужно травить в кипящей смеси кислот. На форуме видел, "шпециалисты" обсуждали, никого не смущала 70% солянка. Будучи новичком не понял где правда, вот и загнул 70%. Прошу прощения. И спасибо за поправку. Ранее читал что 40% это уже геморрой знатный при хранении.
-
Вот! То есть пока на Вюрце температура ниже 110°С - значит выпаривается вода с небольшим содержанием HCl ? B как только поднимется на 110°С и выше - значит осталась азеотропная смесь? В разных источниках пишут что долго хранится под притертой крышкой может только эта смесь, более высокая концентрация проблемна в хранении (в холодильниках или в запаянных капсулах). То есть если мне надо более высокую концентрацию, то её надо порционно готовить перед использованием? Видел идею как капельной воронкой 20% HCl капают в H2SO4, которая отнимает воду, а HCl испаряется (затем в Дрекселя барботированием повышает концентрацию другой части 20% HCl). Это реально? А как-то можно очитстить азеотропную смесь? Удалить всякие примеси, ПАВ? Чтобы чистая HCl*H2O получилась? Или просто сменить колбу-приемник когда начнет перегоняться азеотропная? Прошу прощения за свои "70%". Увидел где-то на форуме и подумал что я совсем не понимаю что такое концентрированная HCl.
-
1. Самостоятельно приготовить растворы для травления цветных сталей и сплавов. 2. Создать и испытать долгоживущее "жидкое олово". Химическое лужение медных дорожек печатных плат перед нанесением паяльной маски. Везде требуется концентрированная солянка.