ТоптуновПотапов
-
Постов
436 -
Зарегистрирован
-
Посещение
Тип контента
Профили
Форумы
События
Весь контент ТоптуновПотапов
-
Этот фрагмент из какой книги? интересуют источники информации [332], [214] и сама, хочу более подробно почитать. Текст нашел тут http://www.chemical-analysis.ru/analiticheskaia-khimiia-azota-spravochnoe-posobie-dlia-khimikov/analiticheskaia-khimiia-azota-0103.html сама книга https://djvu.online/file/ksjdwbCaNsOum Аналитическая химия Азота . Волынец. страница 103 ссылки упомянутые в статье ведут к [332] - РЖХим 1967 20Г68. [214] - РЖХим 1969 4Г103 Если кто-то знает где в интернете найти эти журналы РЖХим с нужными "координатами" подскажите. Такие данные через яндекс плохо "гуглятся"
-
Встречается в химреактивах аскорбинка "для лабораторного применения". Обозначение на этикетке BP2001/USP32 Поверхностный гуглопоиск ничего информативного не даёт. Т.е. непонятно ни происхождение ни предназначение. Только то, что заявляет продавец - для лабораторного примеения. Но а чем она отлична от пищевой? Прошу подсказать, что это за аскорбиновая кислота и для чего. Как расшифровывается обозначение. Почему-то уверен тут сфера проименения/свойства. По свойствам - более слабый восстановитель, чем пишевая. Применяю в методике ПНД-Ф по поределению фосфатов в воде.
-
Благодарю за оперативные ответы и отклик, боле-менее понятно куда копать.
-
aversun, откуда у Вас такие энциклопедические знания обо всём? завидую белой завистью. Да, колонка с амальгамой бы подошла, но хочется применять сложившуюся методику без существенных изменений. В целом она безпроблеманая, кроме цинка. Может его как то активировать можно. Скажем облить кислотой (обнажить поверхность)/промыть дистиллированной водой и применять сразу после активации. Как то так. Пока думаю-размышляю что сделать. Нужен какой-то восстановитель для нитрата в нитрит. Но не слишком сильный, чтобы он не переходил в аммоний. Может быть кислую среду для реакции надо создавать слабой кислотой (в методике, изначально, уксусная). Сейчас приименяю серную, цветность примерно одинаковая, т.е. не дает разительно отличающегося резултата.
-
Для определения нитратов окрашиванием N-(1-нафтил)-этилендиамином используют восстановление нитратов до нитритов (которые собственно и окрашивают) цинком в кислой среде. Цинк применяется в виде порошка типа ПЦР-1 или ПЦР-0 (крупность). Но вот загвоздка, порошки разных партий по разному восстанавливают и цвет получается разный. Некоторые порошки, почему-то, дают вообще в 10 раз отличающуюся окраску, что неприемлемо для количественного анализа. Вопрос - можно ли заменить цинк на магний? или что порекомендуете для восстановления нитратов в нитриты.
-
Благодарю за информацию и вектор деятельности.
-
Извиняюсь за археологию, но чтобы не плодить дублирующие темы спрошу тут: Пытаюсь растворить (приготовить расмтвор молибдата аммония для аналитического определения фосфат-иона) в воде. А он мягко говоря там плохо растворяется. примерно 70% растворилось, а около 30 в виде пыли плавает уже 2 дня. Размешиваю магнитной мешалкой. Подогревать пробовал, растворимость растет не заметно, если вообще растет. В 50 мл не могу растворить 3 грамма молибдата. По идее растворимости хватает, но что-то я делаю не так. Реактив "древний" с хранения, лежит с 2011 года. Что с ним можно сделать, чтобы привести в чувство?
-
не не, это не наш метод. В химреактивах есть нормальный "бирюзовый" железный купорос как минимум ЧДА квалификации. без всяких примесей и трехвалентных составляющих.
-
Ну, допустим. Логика есть, но по опыту скажу что пробка из колбы на кипящей бане тоже не вылетает. При кипящей воде в бане, в колбе вода никогда не кипит. Да бох с ней. Идея понятна, благодарен за комментарии.
-
Нашел какую то методику в этих ваших интернетах. Диктую, кто хочет, может записывать: Берется сухой препарат глюконата кальция и смешивается с сульфатом железа (II) 7 водным. На 100 грамм глюконата берется 64 грамма железного купороса. С12H22CaO14 + FeSO4*7H2O = C12H22FeO14 +CaSO4*2H2O Мм CaGlu = 430,4 г/м Mm FeSO4*7H2O = 278 г/м Смешивается и сразу помещается в колбу которую нужно заткнуть пробкой чтобы глюконат железа не окислялся кислородом воздуха. В колбу добавляют 100 грамм воды и ставят на водяную баню при температуре 70С Через 60 минут, когда реакция завершится, реакционная масса разделится на 2 слоя. Внизу выпадет сульфат кальция двухводный (гипс) а в водной фазе останется глюконат железа. Для разделения испоьзуется горячее фильтрование на воронке бюхнера. Гипс остается на фильте, фильтрат собираем в делительную воронку. Полученный раствор заливаем равным обьемом изопропилового спирта, перемешиваем и оставляем до разделения жидкой фазы на 2 слоя. Нижний, содержащий глюконат, сливается в колбу. Верхний можно выпить утилизировать (можно перегнать и использовать повторно) Нижний слой снова разводится изопропиловым спиртом того же обьема и тщательно перемешивается. В осадке - глюконат железа. Постепенно глюконат полностью из спирта выпадает и его нужно отжать. Например на центрифужке или фильтр-прессе. На худой конец просто выложить в открытую тару (кристаллизатор) и высушить от спирта без отжима. К методике вопрос - почему на водяную баню при 70, а не 95-100? Сульфат кальция с ростом температуры растворим все меньше, по имдее выделить его полнее можно при бОльшем нагреве.
-
Либо на DTPА кислоте (комплексоне) Кто подскажет, как правильно приготовить Fe-DTPA. DTPA есть.
-
Мне в аквариум. Глюконат железа в виде раствора продают многие "дорогие" бренды по аквариумной тематике. Например seachem fluorish стоит 5000 рублей/литр. В литре 10 (десять) грамм железа. Ахтунг! Препарат - глюконат железа продается в любом химмаге 1000-2000 рублей / 250 грамм (4000-8000 р/кг). Купить, технически и практически, могу, хочу ознакомится с методикой изготовления. Может быть там вообще "как два пальца". Внутрь принимать не буду, цели другие
-
Подскажите литературу на методику получения глюконата железа. Купить я знаю где, но как-то кусается, и он портится довольно быстро. (за пару лет, ага)
-
Полностью с этим утверждением согласен. В открытом сосуде можно при атмосферном давлении жечь любой порох, не сильно опасаясь взрыва, проблемы начинаются если сосуд не похож на открытую чашу, а похож, к примеру, на пушечное ядро. Тут уже показатель степени решает.
-
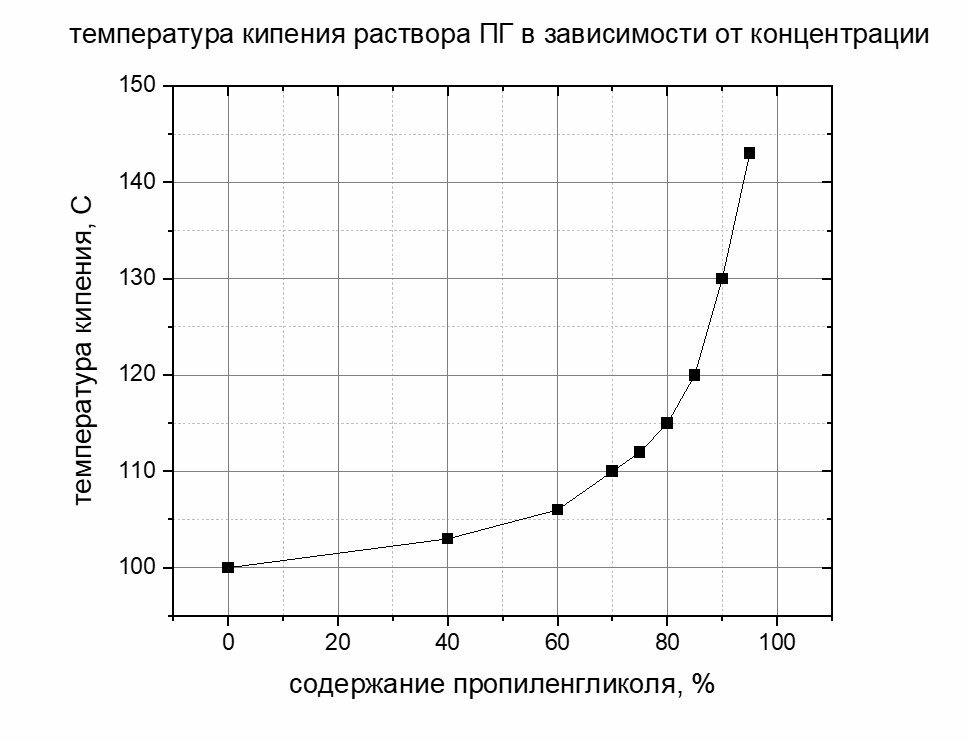
Если вдруг кому то тоже нужна информация по пропиленгликолю, то есть "A Guide to Glycols" компании dow chemical. гуглится по парномеру файла 117-01682-01-a-guide-to-glycols.pdf В документе на последних страницах есть информация о температуре кипения растворов при разных давлениях, в том числе при 760 мм рт.ст. Из этого графика "стырена" температура и концентрация ПГ в жидкой фазе и построен свой график. Не претендуя на высокую достоверность и качество исполнения привожу его как есть.

-
По каналам - прогрессивное горение, по наружной поверхности дегрессивное. После воспламенения наружные слои, сгорая, уменьшают площадь горения, а каналы разгораются и площадь горения в каналах растёт. Одно другое компенсирует в различной степени, но в целом массовая скорость выравнивается (+-). Можно всё детально посчитать, вопрос нужны ли эти выкладки тут? Совершенно очевидно. Уравнение Бернулли для истечения газа. V = (2*(P-P0)/ρ)0.5 Разглядывая уравнения с разных сторон, применяя математическую логику, видно, что истечение газа пропорционально корню из давления. А газоприход пропорционален давлению в степени ν. Для порохов с коэффициентом ν более единицы равновесие между исчечением из полузамкнутого обьема (газорасход) и газоприходом достигнуто не может быть принципиально, потому что газоприход с ростом давления растет быстрее, чем растет газорасход. Наступает классическое ПГД - преход горения в детонацию.
-
Конечно там зависимость, в строгом смысле, далека от линейной. А зависит от хреновой горы факторов. Более того, существуют катализаторы, которые показатель степени "ню" в определенном интервале давлений делают отрицательным и скорость горения не только не увеличивается с ростом давления, но и наоборот - уменьшается. в Рамках этого топика нет надобности лезть в химию, технологию и процессы при горении специальных видов порохов. Для традиционных пироксилиновых зависимость весьма простая V=bP^а. Р - давление, b - константа зависящая только от марки пороха (единицы), а - для порохов = от 0 до 1. Если показатель степени больше 1 - порох увеличивает скорость не линейно. такие пороха не находят применения ибо горят с ускорением, что, очевидно, переводит горение в детонацию. степенная, да, но степень меньше 1. Обычно 0,5-0,7
-
Где бы в литературе найти график или табличку связывающую температуру кипения водных растворов пропиленгликоля с его концентрацией. Помогите, пожалуйста. По этиленгликолю полно, по пропиленгликолю что-то не могу никак найти.
-
не не не. вы путаете массовую и линейную скорости горения. Ща поправим. Чем меньше калибр ствольной системы тем меньше толщина горящего свода. Толщина горящего свода - это толщина самой тонкой части порохового элемента деленная пополам. Уменьшение толщины горящего свода ведет к росту массовой скорости горения (грубо - больше площадь горения. Но не только). Любой порох, хоть в ракете, хоть в стволе гаубицы, хоть в стволе винтовки горит со скоростями одного порядка - несколько десятков мм/сек. Скорость газоприхода можно обеспечить увеличением площади поверхности горения. Опуская нудный вывод о связи массовой скорости горения с толщиной горящего свода и скоростью газоприхода просто приведу факт: чем меньше калибр орудия, тем меньше пороховой элемент. При сохранении одинаковой скорости горения, к примеру 100 мм/сек пороховой элемент в виде зерна с размером 500 мкм сгорит за... 0,5/100 = 0,005 сек. а если в нём сделать сквозное отверстие, то уже вдвое быстрее, не смотря на то, что габариты остались прежние. Длина ствола, к примеру, АК образца 1975г = 415 мм. Дульная скорость пули (на дульном срезе) 745 м/с. Значит теоретическое время полного сгорания пороховых элементов 0,415 / 745 =0,56 мсек. (на самом деле в 2 раза дольше - пуля не сразу приобретает такую скорость, а разгоняется от 0. Поэтому время ~ 1 мсек. Но на понимание не влияет, привожу гипотетический расчет) Допустим у нас порох со скоростью горения 100 мм/сек (это не много и не мало, таких порохов вагон) Значит толщина горящего свода для порохового элемента = 100*0,00056 = 0,056 мм. достаточно сделать пороховой элемент размером 1 мм а в нем наделать 5-7 каналов и получим нужную толщину горящего свода, а отсюда и малое время горения. Итого при одинаковой скорости горения можно одинаково эффективно применять одни и теже пороха в разных калибрах. На фото классический семиканальный пороховой элемент от патрона 7,62х39 (автомат калашникова) Для какой-нибудь пушки, типа известной по репортажам ОРТ гаубицы "гиацинт-Б" длина ствола составляет 8197 мм, скорость снаряда 945 м/с. время горения пороха в стволе = 8,197 / 945 = 8,7 мсек. Толщина горящего свода при линейной скорости 100 мм/с = 100*0,0087 = 0,87 мм. т.е. можно сделать трубку (макаронину) с толщиной стенки 1,8 - 1,9 мм. Они, собственно, так и выглядят как трубки желтого цвета (нитроцеллюлоза техническая - желтая) Так же необходимо помнить, что в ствольных системах давление газов составляет тысячи атмосфер, а в ракетной технике (твердое топливо) на уровне 70-100 атмосфер (в среднем) Скорость горения зависит от внешнего давления и представляет собой, в традиционном представлении, прямую линию - зависимость от давления линейная. поэтому даже взяв один и тот же порох можем получить в 5-10 раз отличающуюся скорость для пушки и ракеты.
-
это, простите, в каких марках порохов наблюдается скорость горения исчисляемая метрами в секунду? Как же, тогда, ракета (двигатель) может работать сотню секунд. Там что, слой пороха несколько километров? Нет, вы ошибаетесь. Как инженер по порохам и ТРТ сильно сомневаюсь, что такие скорости достижимы в принципе. И нигде, кроме специальных средств ПРО (последний рубеж) нигде не применимо. Обачно скорость горения пороха на основе пироксилина (бездымные всех марок) составляет единицы мм/сек. Что в манометрической бомбе вырастает до нескольких сотен миллиметров в секунду, но в любом случае не метры. В баллиститах завести детонация можно, но она там неусточива(я). и применять так же "легко и просто" как какой-нибудь классический гранулит на основе аммиачной селитры не получается. - полно отказов.
-
Я догадывался, хотел спросить коллег на форуме. Вы подтвердили, что способов дельных, лежащих на поверхности нет. Будем думать. Очень благодарен за дискуссию и накиданныые варианты, даже с учетом что их не много.
-
среднерыночная стоимость ПХА ~700-1000 руб/кг в зависимости от его параметров - размера частиц и чистоты. в 300 литрах раствора ~ 65 кг ПХА. в реальности раствор после перекристаллизации примерно 1500-2000 кг ПХА уже мало пригоден для его дальнейшей эксплуатации. Идет брак по продукту, он загрязнен натрием. Хочется способ который позволит натрий выделить, раствор использовать дальше.
-
пользы никакой нет, один вред. Вносится как один из продуктов в реакции. NaClO4 + NH4NO3 = NH4ClO4 + NaNO3 ПХА в осадок, отжимается на центрифуге, далее перекристаллизация. Со стадии получения в кристаллах остается нитрат натрия в виде раствора. ПХА мелкий, остаточная влажность как не жми ~ 4,5% В этом растворе ~ 700 г/л нитрата натрия. т,е в 100 кг продукта 4,5*0.7 = 3кг NaNO3. это ОЧЕНЬ много. На стадии перекристаллизации снижаем до 0,5% max, далее раствор в утиль. Вот "раствор в утиль" очень накладно (дорого) Хочется натрий убрать/заменить на любой ион не дающий окраску пламени.
-
Очень интересная полемика развернулась. Идея с электролизом на ртутном катоде в целом мне понравилась. Но опыт подсказывает, что будет лететь аммиак. у него потенциал ниже (-2,7 у Na/Na+ против -0,5 В у NH3/NH4+) т.е. он начинает выделяться существенно раньше, чем натрий. Но идея любопытная.
-
Добрый день. Есть необходимость удалять ион натрия из насыщенного раствора перхлората аммония. Обьем раствора ~ 300 литров. Раствор используется для перекристаллизации перхлората аммония. (выращивание и окатывание кристаллического ПХА) в качестве примеси в раствор вносится нитрат натрия. Немного, но всё-таки есть. Постепенно накапливается и раствор становится не пригодным для перекристаллизации (при отжиме на центрифуге остается маточный раствор, в котором есть натрий и он попадает в продукт после сушки, отжать досуха естественно центрифуга не может. Нужна ионообменная смола которая заменит ион натрия на, например, на ион водорода или аммония. Концентрация натрия ~ 10-20 мг/литр. Регенерация желательно в кислоте. Подскажите где посмотреть, почитать, может быть есть готовые решения. Заранее благодарю.