

SilverKay
-
Постов
479 -
Зарегистрирован
-
Посещение
-
Победитель дней
11
Тип контента
Профили
Форумы
События
Сообщения, опубликованные SilverKay
-
-
-
Как можно узнать оксид ли там марганца вообще? (в батарейках в смысле)
Только что попробовал прокалить черную штуку из батареек.: взял ровно грамм её, калил тщательно до красного коления, летело немного искр, после прокалки смеюза потеряла в массе 25%, тоесть 750мг осталось. Решил сделать из неё термит, взял около 300мг алюминиевой пудры и смешал, на кончике плоской отвертки зачерпнул смесюги и хотел испытать. Грел газовой горелкой, смесь раскаляется до красна но не горит. Может температуры мало у горелки?
-
С перекисью пробовал, правда перекись 3% и кислота 36%. Шипит, но оно и без кислоты шипит если на черную массу просто перекисью капнуть. MnO2 является катализатором разложения перекиси. Но сульфата я так и не увидел. Может не те условия реакции? Кислота у меня не концентрированная была, но она концентрировалась по ходу варения смесюги. Даже сернокислый туман уже пер, а черная масса так и не растворялась.
Пробовал даже растворить совсем небольшое кол-во черной массы в предварительно упаренной серной кислоте до белых дымов. Прямо не снимая с огня варил в серной кислоте совсем небольшое кол-во черного порошка из батареек. Результат нулевой. Ради прикола решал профильтровать. Бумажный фильтр моментально разъело.
-
День добрый. Решил я попробовать сделать сульфат марганца (II). Реакцию нашел в справочнике по химическим свойствам неорганических веществ.

Что сделал я: добыл из батареек черную массу, батарейки солевые были, насколько я понял. Отмыл черную массу от угля, дополнительно прокаливать не стал. Взял электролит, добавил черную массу в стехиометрической пропорции. Парил-парил, как черная грязь была в стакане так и осталась. Решил разбавить водой и отфильтровать.
Вот что осталось на фильтре(осадок на столько черный, что под каким углом не фоткай - будет черное пятно в центре экрана):
Фильтрат был оранжевого цвета, я его выпарил и вот что получилось:
Уж не знаю что это, но это явно не красно-розовые кристаллы. Эта оранжевая гадость при упарке пахла серой, причем не как серная кислота воняет или спички, а приятный такой запах серы, как у горящей комовой серы.
Итак, вопрос: что за говно у меня получилось? Как перевести MnO2 в MnSO4?
-
-
А что тогда летит при упаривании электролита?
-
Извиняюсь за поднятие старой темы, но до тех пор пока источником серки будет являться элик, эта тема всегда будет актуальна.
В основе идеи о получении 98,3% х.ч кислоты из электролита, которую я и хочу с вами обсудить, лежит выдержка из книги о чистых хим. веществах знаменитого Ю.В.Карякина:
Зеленым маркером я выделил наиболее интересные моменты, которые мне приглянулись. Если я правильно понимаю то, что описано в этой книге, то для того, чтобы получить 98% серку, надо парить элик не до белых дымов, а до момента прекращения их выделения. Автоматически встает вопрос, а куда все эти белые дымы девать? Во-первых это опасность для окружения, а во-вторых это потери. Решение этой проблемы читается далее по тексту, где автор рассказывает про перегонку. Но тут стоит обратить внимание на тот факт, что не уточняется какой концентрации кислоту можно парить в колбе Вюрца, но я предполагаю, что Карякин имеет ввиду все таки конц. серную кислоту, а это очень важный момент в плане безопасности. Все мы знаем что надо лить кислоту в воду,а не на оборот. И если подвергнуть перегонке электролит, то сконденсированная на горле колбы вода может капнуть в кислоту и привести к нежелательным последствиям. Я предполагаю, что именно по этому электролит выпаривают не в колбах, а в стаканах, банках, гусятницах, кастрюлях и.т.д., а вся эта посуда имеет горло, как минимум, не уже дна!
Итак, ближе к делу. Получение азеотропа 98,3% я предлагаю провести в 2 стадии. На первом этапе мы выпариваем электролит классическим методом в химическом стакане ровно до появления белых дымов. У нас должно получится что-то порядка 75-90% загрязненной пылью из воздуха кислоты. Полученную кислоту я предлагаю разделить на 2 равных части. Одну часть мы помещаем в колбу Вюрца или круглодонку с насадкой Вюрца и добавляем туда хромовый ангидрид ровно по Карякину, вторую мы переливаем в колбу-приемник. После того, как кислота с хромовым ангидридом вполне отстоится, мы начнем перегонку. Вся фишка в том, что я хочу растворять SO3 который попрет из колбы Вюрца через холодильник в кислоте, находящейся в колбе-приемнике. Так как процесс растворения идет с выделением тепла, то нам надо позаботиться об охлаждении приемника. Как только белые дымы перестанут переть, мы меняем колбу приемник на чистую колбу и отбираем уже азеотроп - этот процесс проводим согласно Карякину, и по его же словам, в приемнике мы уже будем иметь 98,3% серную кислоту квалификации х.ч. А вот в первую колбу-приемник мы добавляем хромовый ангидрид - это сырец для следующей перегонки аналогичным образом. Извиняюсь за много буков, но процедура не безопасная и лишняя подробность тут совсем не лишняя. Что скажете, господа?
-
 2
2
-
-
Приобрел рН - метр с данным электродом, и появилась пару вопросов. Первое: в чем его хранить? в растворе 0.1M HCl, в растворе 3M KCl или в чем? Все что написано в документации про это "между измерениями рекомендуется хранить в растворе 3М KCl", но "перед вводом в эксплуатацию выдержать в 0,1М HCl не менее 8 часов" А НЕ между измерениями и после отмачивания в солянке в чем?. Второе: на дне синего стеклянного шарика плавают какие-то черные частички, это AgCl? А плавают, чтобы раствор всегда был насыщенным? Третье: непонятен из документации термин "истечение электролита" - это вообще что? Заранее спасибо за ответы, до этого опыта с рН-метрами не имел совсем, да и не химик по специальности=)
-
8 мм, но это на шариковом(всегда думал, что входы-выходы стандартные).
Спасибо за самый адекватный ответ, вопрос снят=)
-
Вы не ответили на мой вопрос: он (этот холодильник) имеется у вас на руках, или вы подбираете на глаз шланг,т.к. ваш холодильник на пути к вам?
Холодильник и не на пути ко мне,и не у меня. Я его в руках не держал, но речь идет о тех холодильниках, которые продаются в русхиме. Давайте для конкретики возьмем, к примеру, вот этот http://rushim.ru/product_info.php?products_id=609 ХПТ-1-100-14-14.
-
Спасибо за столько советов как надеть шланг, но я спрашивал не как надеть, а какого он диаметра должен быть? У кого есть холодильник Либиха? просто скажите какого диаметра шланг вы к нему подключаете(был ответ 10мм), есть еще варианты?
-
Разве входы бывают разными? Почитал ГОСТ 25336-82, но там не указан размер входов и выходов воды. А как удаленно подобрать шланг к холодильнику не знаю. Был бы он у меня, я бы померил.
-
Для подачи воды в холодильник Либиха, какого диаметра шланг используется?
-
Кличко, ты?
-
-
-
CuO я уже получал=)
-
Осадок подсох, красивый, даже жалко его в купорос перегонять, можно его оставить для коллекции синтезированных реактивов:
-
Итак, как и обещал, показываю как азурит в малахит перешел:
Легенда гласит, что когда Глаубер исследовал сульфат натрия, он нечайно оставил эту соль на кухне и кухарка готовя еду перепутала эту соль с обычной. В итоге гости почувствовали "окрыляющий" эффект трапезы и мигом создали очередь к колозету(а может кто и обос*ался в итоге, но легенда об этом умалчивает) В википедии другая история открытия, но не суть. В каналью раствор всегда успеет, я решил с ним поэкспериментировать. Сам раствор я декантировал, профильтровал, долил расчетное кол-во серки. В итоге получилась очень забавная штука: часть CO2 выделившаяся в процессе реакции, растворилась в растворе сульфата натрия и мы волей-неволей получили "газировку от запоров":
Стоит эту колбу взболтать как шипеть начинает с новой силой - аналогично обычной газировке.
Ну а помытый осадок сохнет на фильтре и ждет своей участи:
-
В соль экстра добавляют антислеживающие добавки, что бы она в камень не превращалась.
-
-
Стакан до сих пор стоит, я еще не мыл осадок, но сейчас идет превращение азурита в малахит, не смотря на то, что соды был избыток более чем трехкратный, завтра сфоткаю - покажу. Думаю, разницы нет: из малахита купорос получать или из азурита. Для малахита, думаю, формула будет такая: CuCO3*Cu(OH)2 + 2H2SO4 + 7H2O → 2CuSO4*5H2O + CO2↑. Раствор Na2SO4 c остатками соды, думаю, донейтрализовать серной кислотой после декантации с малахита, профильтровать, и попробовать кристаллизовать Na2SO4 - жалко мне какой либо сульфат в каналью сливать.
-
Хорошо, а когда я буду заливать полученный осадок кислотой после помывки, он полностью растворяется или должно что-то остаться? Вы формулу взаимодействия азурита с серной кислотой помните? Если кислота будет не 40%, а 36%(элик 1.28 г/мл), ничего страшного? Как-то неохота на 4% упаривать.
-
Ну допустим:
И на каком же фильтре мыть осадок чтобы не нахватать ворсинок? Почему жидкость наверху синяя? Там же сульфат натрия? Глауберова соль?
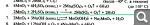











MnO2
в Общий
Опубликовано · Изменено пользователем SilverKay
Звездочёт был прав. Первый раз я дул пламенем горелки прямо на черную массу и сжигал много кислорода. Я провел прокаливание еще раз с новой порцией, но уже держа в уме старые грабли. В этот раз я направлял струю огня не внутрь тигля а раскалял его дно:
Когда вся масса стала светится красным светом я периодически вынимал тигель из-под огня, интенсивно перемешивал угли медной проволочкой и одновременно их раздувал:
В процессе прокалки никаких искр не летело, пламя непосредственно на препарат не направлялось. Сабж потерял в массе 30% на этот раз, а учитывая, что прокаливал я предварительно промытую водой массу из батарейки, то изначально там углерода в районе 50% по массе, а может и больше. Ну и наконец-то масса приобрела вожделенный коричневый оттенок, как и положено по справочникам. Хоть камера на телефоне и хреново различает близкие цвета, но все же:
До прокалки:
После прокалки:
Для того, чтобы убедиться, что прокалил весь уголь, провел повторное прокаливание и перевзвесил, масса потеряла еще 0.02г, но это скорее издержки пересыпания и налип на палочке для помешивания. Оттенок светлее не стал.