the_Rion
-
Постов
1275 -
Зарегистрирован
-
Посещение
-
Победитель дней
1
Тип контента
Профили
Форумы
События
Весь контент the_Rion
-
Вооот, сульфат натрия это уже намного больше на правду похоже. Это наверное вот что, это этилсульфат натрия загидролизовался. Тем более его двойное количество было, поскольку я парацетамол зажлобил много его тратить, он мне еще от головной боли понадобится. Думаю надо бы эти кристаллы хлоридом кальция проверить, будет ли гипс сыпаться, или нет. А вообще надо было мне конечно сперва едкий натр в спирте растворить, потом уже к нему все остальное кидать. Но я думаю что надо все-таки через уксусный ангидрид делать, из фенетидина.
-
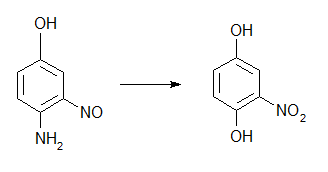
Вот я смешал этилсульфат натрия в этаноле, парацетамол и едкий натр, потом прокипятил полчаса слил с осадка и разбавил водой, в результате ничего не выпало. Осадок был идентифирован как едкий натр, гранулы которого не растворились в этаноле. Следовательно логично что фенацетин не выпал. Тогда я кинул едкий натр в полученный мною раствор воды, спирта и прочей херни, т.е. парацетамола, этилсульфата, в результате чего этот раствор почернел, чего и следовало ожидать. Тогда я подумал - сука блять, ну как же так, надо было не быть дебилом, всего-то нужно было взять весь этанол и продегустировать, как обычно, все-таки я выпивал этанол быстрее чем делал его из браги... Короче я подумал что было бы неплохо попробовать отогнать этанол назад из всей смесюги, то есть я сохранил весь этот черный раствор и постоял он у меня в банке наверное двое суток. Постоял и постоял, к этому времени я решил просто купить денатурат, ну это чтобы у меня был этанол для реакций, а то из браги когда у меня этанол появляется, какие тут нахер реакции... Вобщем слил я из банки черную бурду и нашел на дне прозрачные кристаллы, грязноватые немножко, но холодной водой отмылись, то есть это вещество не растворяется в воде, ну, по крайней мере в холодной. Быть парацетамолом оно я так думаю не может, поскольку едкого натра было более чем достаточно, на этилсульфат натрия это тем более не похоже, сульфат натрия из-за гидролиза этилсульфата или действием едкого натра тоже думаю что нет - воды я к этанолу добавил достаточно, сульфату было бы где раствориться. Этилсерку я гасил кальц. содой напрямки в этаноле, без воды, вся сода погасилась, я потом слил этилсульфат с осадка сульфата натрия в этаноле... Вобщем вопрос такой : Что за гавнище получилось, почему эти кристаллы прозрачные (после промывки водой) если это фенацетин, и чего он тогда в водном растворе тоже образуется что ли? Чего-то хня какая-то получается, я то думал он только в этаноле получится, а вода уменьшит выходы или вообще не будет с водой получаться, не? Вообще везде фенацетин описывается как белые кристаллы, в одном только Беркенгейме говорится что кристаллы прозрачные.
-
 Вот-вот, это самое неприятное. Поэтому NO2 я стараюсь избегать по мере сил и возможностей. У хлора, например, такого эффекта имхо нет. Помню передышал хлором как-то, было жжение в носу и в горле, ну недолго, полдня где-то и прошло. Раз живой, значит все норм. А с NO2 получается можно травануться и не заметить. Хотя вобщем-то он воняет, не знаю как им надышаться можно, но вангую что он типа принюхивается, если концентрации небольшие. Да все мы когда-нибудь умрем, это точно =)
Вот-вот, это самое неприятное. Поэтому NO2 я стараюсь избегать по мере сил и возможностей. У хлора, например, такого эффекта имхо нет. Помню передышал хлором как-то, было жжение в носу и в горле, ну недолго, полдня где-то и прошло. Раз живой, значит все норм. А с NO2 получается можно травануться и не заметить. Хотя вобщем-то он воняет, не знаю как им надышаться можно, но вангую что он типа принюхивается, если концентрации небольшие. Да все мы когда-нибудь умрем, это точно =) -
Бензол пишут канцероген, вроде как рак вызывает. Так или не так, но я бы им к примеру, мыть руки не стал бы. Вообще я так понимаю, это все при хроническом воздействии. Думаю что если подышали и никто не издох в течение суток, но ничего, бгг... Азотная, ну я с ней без ничего работаю, с азотистой также, без перчаток и респираторов, правда когда нитрозирую, то дверь открываю в сарай чтобы диоксид азота вытягивало, ну как можно ближе к двери работаю, ну, диоксид, конечно воняет, но проблем с ним у меня нет, т.к. он быстро улетает на улицу. Хотя вообще азостистая вреднее чем азотка, возможно что такими методами я лет на 5 раньше издохну, хотя в целом не факт. Ну и еще чтобы без перчаток, это надо чтобы твердая рука была, бгг... Типа как у хирурга., вот. Вообще имхо NO2 не канцероген, однако я слышал что якобы отравление им наступает не сразу... Короче не советую им дышать =)
-
Натрий я видел на Ютубе делал Огненное ТВ электролизом расплава едкого натра, получается недорого, можно попробовать. Сплавлением м-бензолдисульфоната натрия со щелочью это наверное самый простой способ, если есть олеум, хотя может я могу его купить, хз... Есть еще такой вариант, это мета-ксилол, который в принципе и есть почти готовый резорцин, если заменить метиловые группы на гидроксилы, есть даже патент, где синтез сводится к тому, что мета-ксилол сперва хлорируют, далее окисляют до дихлорофталевых кислот и потом гидролизуют в автоклаве в водном растворе едкого натра, но я думаю можно просто сплавить соли этих кислот со щелочью. Патент : https://patentimages.storage.googleapis.com/90/a3/dc/036c21afd17b08/US3903177.pdf Если у меня был бы чистый сульфид, тогда да. У меня смесь сульфида с сульфитом, участвует ли сульфит в восстановлении чего-либо, я не знаю, потому что всегда смешивал столько серы и щелочи, чтобы получилось нужное количество сульфида, а на наличие сульфита я просто забивал и все, это отлично работает при полном восстановлении, а при частичном хз как будет. Поскольку по одним источникам сульфит в некоторых случаях восстанавливает, по другим источникам он якобы вообще не участвует в реакции, всякие там гидросульфиты и дитиониты насколько я понимаю тоже могут восстанавливать, тиосульфат вроде как нет... Так что тут наверное восстанавливать полностью обе нитрогруппы... чтоб было надежнее.
-
Через нитроанизол это тоже что и получение гваякола по Беркенгейму (страница 107), хорошая методика, когда уже есть готовый начальный изомер нитрофенола. Если метилировать через метанолят натрия, это потребует наличия металлического натрия, возможно что можно его купить, только вот хз сколько будет стоить. Либо метилировать метилсульфатом натрия, который не самый лучший метилирующий агент. Либо по Беркенгейму метилирование вести метилтозилатом, который придется делать также самому. В той же мере в Евросоюзе парацетамол слишком дорогой, 1 евро за 10 таблеток минимум, это всего 5 граммов, а значит мне проще вести синтез парацетамола самому из фенола, который у меня есть, хотя это встанет в синтез уксусного ангидрида, и еще вопрос как будет идти восстановление п-нитрофенола сульфидом натрия, я думаю что идти будет плохо, так как прописи обычно требуют вести это дело сероводородом... Хотя имея фенол мне проще получить фенацетин через этилирование фенола и его последующее нитрование, с целью получения фенетидина, его далее ацилируем уксусным ангидридом, потом нитруем. Либо что наверное лучше всего это купить бензол и делать по второй схеме через мета-динитробензол, я думаю можно также восстановить сульфидом натрия начисто (обе нитрогруппы), особенно учитывая что сульфид натрия у меня это результат реакции серы с NaOH, а значит - напополам с сульфитом, но все-таки я думаю что сульфит в данном случае не деятельный как восстановитель, хотя хз, хз.... Впрочем, м-динитробензол можно также восстановить и частично, то есть только одну нитрогруппу.... это тоже вариант, подойдет ли для этого мой колхозный сульфид я не знаю, если сульфит реально не деятельный, тогда должен подойти, если деятельный, тогда надо считать и сульфид и сульфит. Олова и железа у меня увы нет, правда есть еще частный случай восстановления с помощью алюминия в NaOH, но эта метода крайне плохо документирована и результаты не совсем понятны. Также по парацетамолу могу добавить что его этерификация через этилсульфат натрия с едким натром идет в целом плохо, то есть для фенацетина нужен фенетидин и уксуный ангидрид, кроме того, нитрозопроизводное парацетамола возможно что гидролизуется едким натром далее чем только до отвала ацетогруппы, поскольку результат гидролиза этого нитрозопарацетамола имеет запах миндаля, хотя это конечно не дает повода думать что гидролиз валит до нитроанилина, но хз, хз... Хотя, как я уже сказал тут в ЕС парацетамол стоит не 4 рубля, как в РФ, а уже целое евро, так что мне это тоже не вариант, у меня было немного российского, я покупал в командировке когда был, но опять же - много не привезешь =))
-
Аркадий, да тут знаете ли к мета-аминофенолу как раз и нужно прийти еще сперва... Уж из него то резорцин сделать - раз плюнуть. Впрочем исходя из парацетамола можно сделать вот что, причем нитрогруппа 100% будет в мета положении : Всего-то требуется этерефикация парацетамола, например с помощью этилсульфата, если нет, ну хрен с ним, этилбромид должен подойти. Но я думаю что этилсульфат натрия тоже прокатит.

-
Канифоль может быть и есть на блошинке, но вообще я ее не встречал нигде ни разу. Есть в хозмагах олово и прочее для пайки, но там емнип какие-то пасты кислотные, сомневаюсь что там в хозмагах канифоль есть и если даже есть то скорее всего что дорогая, могу глянуть конечно. Лимонная есть стоит 4 евро за килограмм. За эти деньги можно купить литр толуола и еще примерно 1 евро сдачи останется. Да самое дешевое и простое это азеотропная перегонка с чем-либо, что с водой не смешивается. Толуол идеально подходит, так как он не экстрагирует уксус от воды и похоже что с уксусом не смешивается, хотя следы уксуса в дистилляте были но там такой мизер что толуол просто промыть водой и все, короче толуол не расходуется практически, да там будут незначительные потери при промывании и т.д., но это фигня. Гнать тоже легко, потому что азеотропа толуол-вода-уксус не существует, поэтому в зависимости от количества толуола и крепости эссенции (уксуса) фракции могут быть такие : толуол-вода, толуол-уксус, уксус-вода и последняя это чистый уксус, кстати если будет фракция толуол-уксус то она я так понимаю что тоже будет расслаиваиться, как и толуол-вода. Самое главное на мой взгляд это все-таки офигенная скорость отгона фракции толуол-вода, при том что толуол с водой валит гладко и стабильно, не надо мудиться по каплям собирать что-то там, просто тупо ждать окно, а его сложно не заметить, когда собирать следующую фракцию. Короче проще, легче и быстрее чем допустим гнать спиртягу 95% из браги на этой же установке что у меня есть. Ну я на неделе новой попробую смешать еще одну партию 100 на 100 грамм и волью к ней имеющийся кубовый остаток уксуса и прогоню все по полной, гляну какие будут еще фракции и как будет уксус гнаться, но это если время будет на это.
-
Я вот думал взять 95-й бензин и отогнать оттуда легкую фракцию, до 50С например кипит которая, а ее там судя по слухам немногим меньше половины, это конечно не петролейка будет и не галоша, но некий потенциал все же должен быть, но у меня толуол в хозмагах продается свободно, поэтому я так подумал нет смысла с бензином возиться, но если толуола нет, тогда это тоже вариант. Вообще в основном литература советских времен, а на те времена я так смотрю с бензолом очень даже дружили и юзали везде где могли, в том числе и как экстрагент вместо диэтилового эфира, поэтому ясное дело что бензол также пристроили и к уксусу, отсюда как раз ноги и растут у методы с бензолом, а вот толуол я не знаю, был ли доступен в советских лабах или нет... Хотя многие методики честно говоря чрез жопу в книгах оформлены, взять то же нитрование фенола к примеру, есть же способы делать без осмоления через ЛУК, так нет - везде одна и та же куетень в водном растворе и потом пиздец с отгоном о-нитрофенола, столько еботни и такое гавнище в итоге получается горелое, что тьфу, да лучше ЛУК потратить без гемора и перегонок, небось дешевле даже будет, а быстрее в разы, но видимо раньше рабочее время не считали, а фенол тем более, который копейки стоил. Хорошо подвернулась эта заметка с толуолом на глаза, а то это ведь пиздец если через серку с ацетатом натрия делать, такой геморой, столько времени тратить на не пойми что, тьфу, как вспомню - сразу дурно делается.
-
Да вот я про то и думаю, чтобы не прошло окисление аминофенола, пускай и замещенного до гидрохинона азотистой кислотой, то есть : Что нафиг не нужно, поскольку я хочу резорцин сделать, а не гидрохинон. Надо пробовать диазотировать как в Википедии пишут, через гидрохлорид аминофенола в присутствии ZnCl2
-
Мне нужен не краситель, мне нужно вклеить нитро или нитрозогруппу в мета положение. Другой вариант это через 1,3 динитробензол и далее мета-нитроанилин. Но поскольку в аптеках есть уже готовый парацетамол.... Правда я не сильно уверен в последнем шаге, как бы амин не издох и не нитрозанулся в процессе, хотя этот шаг вовсе не последний, если подумать. Но там уже вопросов в принципе и нет, все и так понятно. Есть японская работа и она гласит что в среде ЛУК и NaNO2 нитрозогруппа встанет в мета позицию. Нитрозирование я уже провел, при этом достаточно даже и эссенции 70%, смолы не обнаружено. Гидролиз с NaOH также валит быстро и достаточно гладко, правда я не грел, но есть думаю кстати еще шанс на гидролиз вплоть до м-нитрозоанилина, хотя я в этом сомневаюсь. Хммм, у меня же есть бензойная кислота.... Ее нитруем и перегоняем в НБ, далее идем дальше до 1,3 ДНБ, не?
-
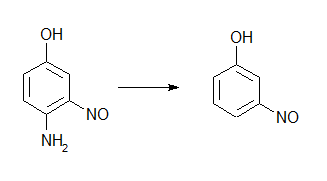
Для этого парацетамол нужно нитровать. А в мета положение он нитроваться не будет. Если только его в среде ЛУК не нитровать, но здесь я тоже хз как процесс мог бы пойти. Поэтому вопрос том, встанет ли нитрозогруппа (не нитро, а нитрозо) в мета положение при заданных условиях, или нет?
-
Как думаете, встанет ли нитрозогруппа в мета положение? Условия следующие - NaNO2 в ЛУК. Из второго третье сделать гидролизом с NaOH или KOH. Если все так и будет, как перейти от третьего к этому : Насколько я понимаю, нужно перевести в гидрохлорид, диазотировать и покипять соль диазония с водой? А прокатит ли это, амин ведь все-таки фенольный, как бы он не пронитрозировался? Ну, какие у кого мысли будут?
-
Вполне должно получиться и без толуола, возможно что попробую так сделать, когда закончу опыт с толуолом. Могу сказать что с толуолом есть фишка вот в чем - толуол с водой летит очень быстро, он правда не то чтобы ручьем валит, но пара капель в секунду будет, а это я считаю достаточно быстрая перегонка, мне есть с чем сравнить. Закипает смесь эссенции и толуола также куда быстрее многих других веществ, которые я перегонял. То есть процесс с толуолом очень быстрый, имхо практически идеальный, я лично считаю что гнать толуол вообще удовольствие, ничего не толкается, закипает быстро, кипит ровно, при фракционировании есть четкое окно между фракциями, я про это писал уже. У меня бутилацетат есть, но имхо толуол доступнее и с ним легче, его легче отмыть потом от следов уксуса. Хотя так то и толуол и бутилацетат получаются что возвратные, можно без особых проблем использовать повторно. Надо бы сделать как я говорил уже, еще одну партию и смешать с уже имеющимся отгоном, отделить воду и толуол, потом гнать дальше и посмотреть как это все будет себя вести, что там фракции будут. Еще хорошо бы ареометром померить плотность у кубового остатка, хотя и так понятно что там почти ледянка, правда похоже что продажная эссенция не 70%, а чуть поменьше будет. Просто у меня весы не калиброваны, значит данные по плотности не полностью достоверны, я даже монеты по два евро этими весами мерил, отклонение от 8,5 грамма на монету гуляют, вобщем надо бы ареометром конечно померить плотность. Ну и да, выморозить попробовать, посмотреть как пойдет этот процесс. Руслан, это все звучит здорово, но мне например тогда канифоль надо покупать где-то, сколько она тут в ЕС стоила бы я не знаю, но скорее всего по деньгам это примерно как делать ацетат натрия из эссенции и соды и потом обрабатывать его 96% серкой, это минимум, 90% серку тоже можно, но думаю это так себе вариант, воды уже многовато в ней... Еще на все про все дополнительно работа это сушить ацетат, что медный сушить что натриевый плавить, потом отходы сульфатов еще... Тогда проще купить бензол, насадку Дина-Старка и удалять воду по 100% проверенной книжной методе через бензол, даже это выйдет дешевле, насадка даже окупится, если считать расходы на отходы солей и затраты на кислоты, еще время тоже считать надо, его (время) мне больше всего жаль тратить впустую. Единственное это чисто в научных целях попробовать через канифоль, ради науки. А так я вижу что с толуолом работает, никаких насадок Дина-Старка при этом не нужно, ну, единственное, это толуол надо подготовить, вылечить его от этой черной оспы (тиофены или что там в нем) сперва, и потом желательно перегнать.
-
Вот в мои загребущие руки угодили толуол и уксусная эссенция, поэтому надо было решать что теперь с этим делать. Поскольку толуол немножко "болел", выделял черное гнойное маслице с серкой (100 мл толика на 10 мл 90% серки) при перемешивании, то толуол был отделен от этого масла и потом перегнан. Далее я взял 100 грамм толуола и 100 грамм уксусной эссенции 70%. Плотность эссенции я не измерял, поверив добрым дядям-производителям. После смешивания оба вещества расслоились примерно напополам (может и не в такой пропорции, но в 1 литровой колбе это выглядит примерно как напополам). Была собрана установка : В результате перегонки было собрано ~130-135 миллилитров дистиллята, разделившегося на два слоя примерно 8 к 2 соотношением, где верний слой состоял из толуола, а нижний из воды. В колбе осталось 65 граммов прозрачной жидкости, не имеющей слоев и масса 10 миллилитров которой составила 10,45 грамма. Дистиллят был собран при температуре ~84,6C, после чего температура в насадке Вюрца упала до 72С, но примерно через пять минут начался рост температуры и при 96С перегонка была остановлена, поскольку обьем остатка слишком мал для используемой системы, т.е. осталось примерно 70 мл жидкости в 1 литровой колбе. Я думаю что надо сделать еще такую же партию или две, обьединить с уже имеющейся, и перегнать, посмотреть, будет ли фракция толуола и уксусной кислоты при 104С, и при какой температуре будет гнаться уксусная кислота. Вообще насколько я понимаю, вода от уксуса таким образом действительно отделяется с помощью толуола. Из собранного толуола и воды, толуольный слой с карбонатом натрия не реагирует вообще. Водный слой не пробовал реагировать. Запаха уксуса в отгоне практически не было.
-
С NaOH будет салициловая кислота, с KOH салициловая и п-оксибензойная. (примерно 50 на 50).
-
Аверсан сказал что нельзя окислить MnO2 гипохлоритом в щелочной среде, так оно действительно и есть. Однако как я уже сказал, можно окислить сульфат марганца гипохлоритом в щелочной среде, то есть NaClO + NaOH + MnSO4 За это есть пруф на sciencemadness, которому нет причин не доверять. Более того, гипохлорит без особых проблем в щелочной среде также окисляет хлорид хрома до хромата натрия, я лично это делал, и это работает. Прбовать с сульфатом марганца я не намерен, хотя у меня такой сульфат есть, просто по той причине что будет очень разбавленный раствор, а Белизну туда придется фигачить галлонами, едва ли не канистру Белизны на стакан сульфата, потому что Белизна дрянь в большинстве случаев, а электролизный гипохлорит тоже не подарок, даже если верить книжкам, что якобы электролизом ЕМНИП можно до 5% гипохлорит сделать, так вот - эти 5% это тьфу и ничего, а гипохлорит надо процентов 10, не менее, иначе потом придется кучу воды выпаривать, ее там реально канистра на стакан будет, почему эта хренотень лучше работает с хроматом? Рассказываю, почему - по очень простой причине, после окисления солей хрома гипохлоритом хромат из раствора осаждают медным или цинковым купоросом и получается хромат к примеру меди, который нерастворим в воде и легко отделяется, я такой хромат тоже делал, потом то уже при желании его переводят в хромат калия или натрия... То есть метода в водном растворе по перманганату она рабочая, но Белизну надо не магазинную а техническую для дезинфекции, вобщем, достаточно концентрированную, от 10%, иначе потом от магазинной Белизны будет воды немеряно, а это очень и очень плохо. И я думаю что лучше по старинке плавить диоксид марганца с селитрой и щелочью, вопрос только как дальше работать, я думаю что лучше газообразным хлором, тогда будет хлорид, скажем калия, ну и побочный остаток нитрита калия после плавления еще.
-
Гипохлоритом можно окислить соль марганца, например сульфат, до перманганата, получив при этом разбавленный раствор. Диоксид марганца лучше сплавлять как обычно с KOH и KNO3.
-
Вангую что надо не просто тупо плавить, а еще и всю смесюгу в процессе плавления перемешивать, тогда и выход будет получше. А так в книжках пишут что якобы из хлороформа можно цианид сделать, правда о требуемых для этого услоявиях молчок - ни бе, ни ме.
-
Потому что проще купить желтую кровяную соль у кого-то кто из лабы спиздит ее, и не заморачиваться с грязными продуктами плавления, хотя смотря зачем вообще цианид нужен, если на бензилцианид, то в принципе можно и поплавить мочевину со щелочью, Chemplayer делал и турбулева синь у него даже получалась потом, но что там за выходы по цианиду количественно - хз.
-
Нитротолуолы гнать при атмосферном не советую, будут осмоляться сильно. Вообще от свойств конкретного вещества зависит, ЭГ нормально гонится к примеру, хотя тоже органика, но это поскольку он не подвержен осмолу или окислению при перегоне при атмосферном. Короче, гнать то все можно, но некоторые вещества при атомсферном гонятся куево (в смысле осмоление происходит) или разлагаются.
-
Книга Бытия, глава 5, параграф 4.
-
Да не вопрос. Только ментол лучше покупать. Методика есть в книге Лазурьевский Г. "Практические работы по химии природных соединений", издание второе, стр. 138. Есть также и в первом издании тоже, хз какая страница. Выход ментола составит примерно до 2% масла от сухой мяты, а ментола собственно от масла это до 70% обьемных. Так что советую купить, а если будете выделять, тогда желаю удачи, она очень понадобится, бггг...
-
Я бензин в 20 литровой канистре хранил, но бензина 10 литров. Правда не очень долго, несколько месяцев, а канистру на пол ставил, там холоднее всего. Даже не знаю опасно или нет это, я слышал что типа в пластике бензин нельзя хранить, но мне в России наливали на заправках в пластик, правда не везде. Вообще есть и металлическая канистра у меня 20 литров, может в нее лучше заливать?
-
Допустим, спирты окислять или стирол. Амины окислять точно нельзя, поскольку из-за наличия нитрита будет еще и восстановление идти побочно. Может вообще нет смысла плавить, а просто окислять диоксидом марганца и все, с избытком серной кислоты? Да, будут затраты на серную, но и плав варить это тоже затраты на газ, чтобы плавить, затраты на едкое кали и нитрат. Если работать дальше до перманганата, так вообще получится что дешевле диоксидом. А вообще по окисляющей способности я так понимаю, они идентичны, что манганат, что перманганат? Просто ведь плав то сухой уже получаться будет, его размолол и все, можно хранить. А чтобы его до перманганата доработать, тут надо еще кристаллизовать, сушить потом, хотя чтобы окислять потом придется опять марганцовку растворять в воде. То есть получается самое простое и быстрое это диоксид, манганат чуть сложнее, но зато он будет активнее чем диоксид.